JCO:如何设计儿童肿瘤联合疗法的早期试验,才能让更多患儿受益?
时间:2023-09-14 20:04:24 热度:37.1℃ 作者:网络
儿童和青少年肿瘤领域存在较大未满足临床需求,需要疗效更好、长期副作用更少的治疗方法。抗肿瘤药物联用时(组合疗法)可以起到机制互补、减少耐药的作用,取得更好的疗效并降低单药的毒性。因此,有必要在儿童肿瘤适应证中积极提倡联合疗法的临床开发。
成人抗肿瘤治疗临床开发在过去十年中发生了巨大的变化,逐渐摒弃了传统按部就班的I/II/III期设计,而更多在早期临床中采用I/II期无缝设计,快速进入剂量拓展阶段,以做到快速验证。但儿童抗肿瘤药物领域相对较落后;对于抗肿瘤新药,儿童适应证的临床开发常落后于成人适应证,中位延迟时间长达6.5年。近年来,不少共识文章呼吁加速抗肿瘤药物儿科适应证的开发。因此,随着组合疗法的增多,有必要制定共识,在药物临床开发的早期阶段对组合疗法进行有效的评估。
新药的联合疗法,可能涉及联用标准疗法(SoC),或者另一种新药(Novel-Novel combo)。目标应是快速、有效地评估药物的疗效,同时也没有损害发现安全性信号的能力。从实操上来说需要确定纳入患者的最低人数,以支持下述目标:确定剂量和给药计划,识别早期疗效信号,为做出“继续开发vs放弃开发”的决策提供充分信息。
此外,为了更有效地在开发儿童适应证,应该在临床前研发中更多地采用儿童肿瘤的模型,获得充分的科学证据,来支持儿童中的抗癌组合疗法。
近期,欧洲和美国都立法促进科学驱动的、以患者为中心的儿童抗肿瘤药物开发。以期肿瘤患儿能尽早用上创新、安全、有效的抗癌治疗。高效的儿童抗肿瘤药物研发需要监管的积极参与。对齐科学、监管和支付方对临床试验的要求,确定最低需要的患者人数。此外,让患者组织也参与到药物研发过程中十分重要;这样做可以让患者的声音被听到,并且发现具体的未满足临床需求。
近日,欧美多位学者在 Journal of Clinical Oncology 上发表了题为 Combination Early-Phase Trials of Anticancer Agents in Children and Adolescents 的特别报告,作者们提供了利益相关方的共识意见,并讨论了在开发儿科肿瘤药的早期开发中采用联合疗法时应考虑的要点,同时尽可能介绍了最佳实践。

儿童抗肿瘤药物早期研发的一般考虑
研究人员建议儿科临床评估一种新的抗癌药物时,采用早期阶段无缝试验设计,而不是单独的 I 期和 II 期试验,因为分阶段的试验会提高样本量的需求。早期临床试验包括两个部分:1)剂量探索或剂量确认阶段;2)剂量拓展,在此阶段获得最优剂量、毒性概况、药动学(PK)参数、药效学(PD)效应和抗肿瘤活性的早期信号(表1)。单药或者联合用药的平台试验,有多个平行的试验臂,是一种可能的设计框架,并且具有诸多优点(表2)。
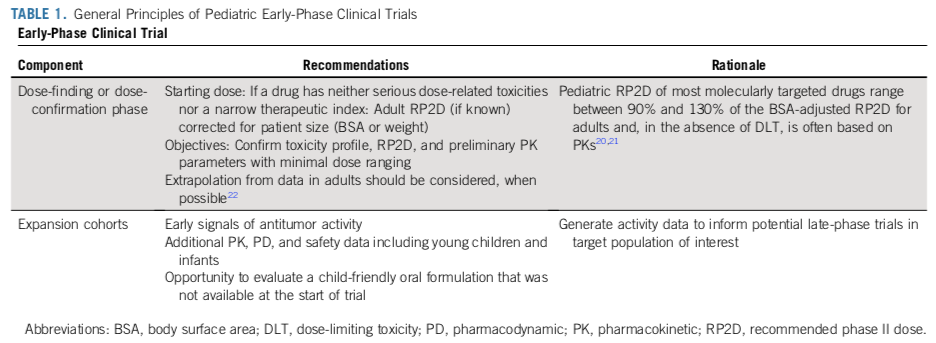

临床实践表明,对于化疗药物而言,12岁以上患者的PK是相似的,因此不需要区分年龄队列进行入组;对于2岁以下的婴幼儿,有必要采集额外的PK/PD数据,因为低龄儿童可能会有不同的药物代谢情况。早期试验应避免在入选标准中对患儿年龄设定下限。开发适合患儿年龄的剂型很重要,但是不应该为了开发特殊剂型而延迟临床试验。
儿童抗肿瘤联合疗法开发的几点考虑
一般原则
在设计联合用药试验时,和单药试验类似的,也应该采取无缝设计,并且包含剂量探索/确认和剂量拓展的阶段。应考虑到对不同药物的先验知识,以采取合适的试验设计。药物组合中可能包含已经在儿童中有临床数据的药物,也可能包含仅仅在成人中有应用经验的药物。联合用药可能带来新的毒性;组合中药物可能具有相似的毒性谱,使毒性叠加放大。对于新药联合SoC化疗的设计,应考虑当下对新药的PK和安全性情况的了解程度,以及是否会有重叠的毒性谱,或者药物相互作用;对于novel-novel combo,具体情境取决于:1)儿童PK和安全性情况是否清楚;2)成人PK和安全性情况是否清楚;3)是否有药代的特殊性,导致药物相互作用;4)是否有重叠的毒性(如表3,图1)。
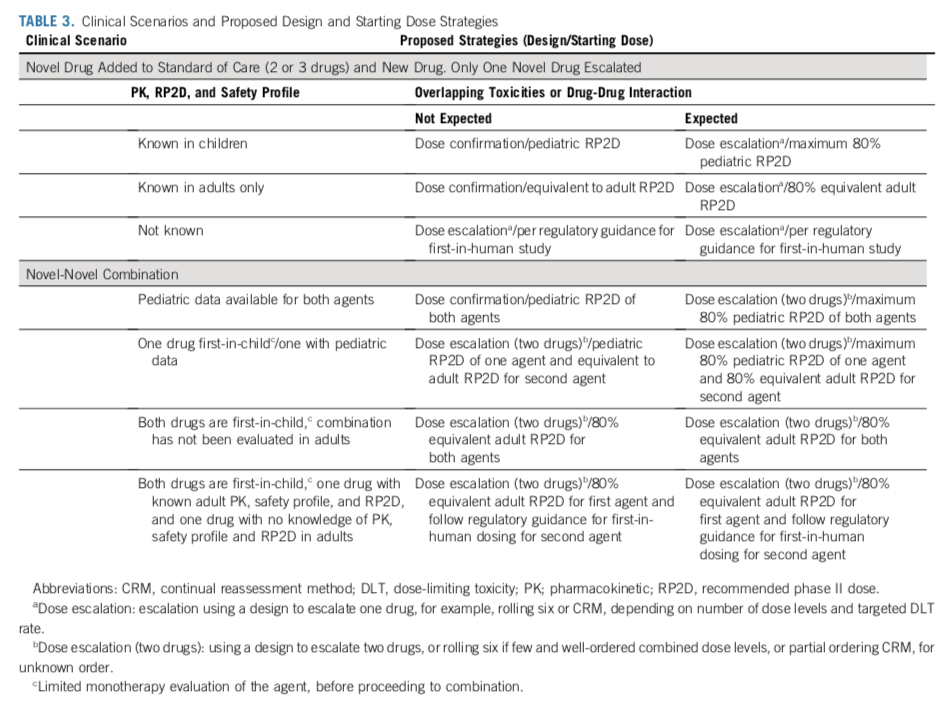
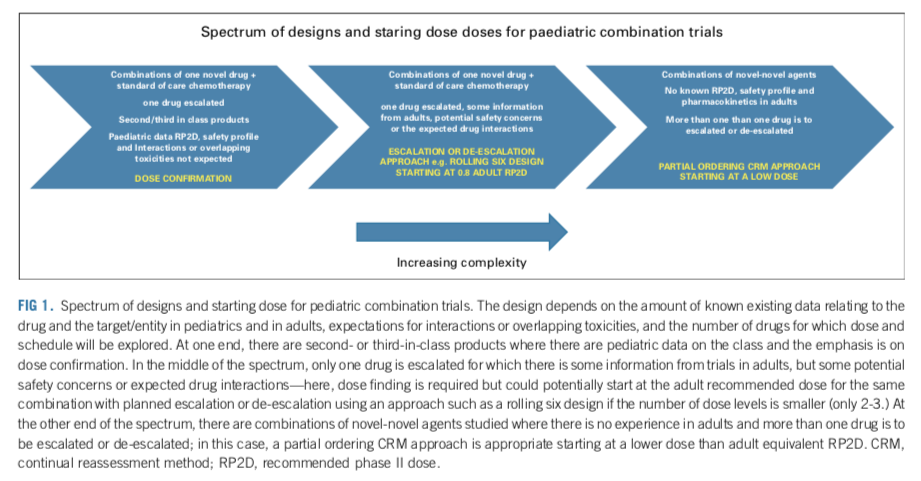
父母让患儿参加临床试验,是希望这项研究中的疗法能让患儿获益。因此,虽然试验的目的是回答科学问题,但治疗的目的也应被纳入考虑。应在剂量探索阶段就有选择地纳入患者而不是纳入all comer,以增加患者获益的机会;避免亚治疗剂量和无效暴露;减少单药治疗的时间;快速评估疗效。为了减少亚治疗剂量的暴露,可以考虑在同一名患者身上进行患者内剂量爬坡。对于联合疗法来说,采用随机对照试验设计,将新药加标准疗法vs标准疗法进行比较,是阐明新药疗效和毒性的最有效方法。平台试验可能对联合研究的评估很有价值。在拟议的临床开发途径中,患者权益倡导者和监管机构应尽早与研究者接触,试验设计必须考虑监管要求。
联合疗法的立项逻辑
对于联合疗法,其立项应有非常清晰的肿瘤生物学背景和临床获益潜能。现在越来越多例子显示,单药没有疗效,但是将药物和标准治疗连用,有临床获益,比如利妥昔单抗用于B-NHL,三氧化二砷用于APL,以及博纳吐单抗用于治疗B-ALL,等等。
非临床产生的证据至关重要,其目标是组合药物的活性至少具有相加性或协同性,但要注意对正常组织的毒性问题——药物组合是否有足够的安全窗?因此,需要进行体内研究,并进行适当的对照。例如,O6-苄基瓜氨酸加亚硝基脲类治疗高级别胶质瘤:临床前观察到该组合组合具有显著的协同作用, 但在患者体内,对正常造血细胞也发生了协同的药效增强,导致过度的骨髓抑制,因此在给予 O6-苄基鸟嘌呤时,需要减少亚硝基脲的剂量。一项 III 期试验比较了 O6-苄基鸟嘌呤加减量双氯亚硝基脲、卡莫司汀(BCNU)和全剂量 BCNU 治疗成人高级别胶质瘤的效果,但因无效而提前终止。
支持联合用药所需的临床前研究的广度和深度,取决于基本生物学假设的稳健程度。例如,如果拟纳入组合的是针对一个明确的癌症驱动基因的靶向药,那么所需的证据可能较少。相反,如果缺乏预测疗效的强生物学逻辑和生物标志物,那就需要有强有力的临床前证据来证明其具有实质性的联合效应。ITCC-P4 和儿科临床前试验联盟(Pediatric Preclinical Testing Consortium)就临床前试验的最低要求达成了共识,以促进研究疗法的开发。
试验设计考量
入选标准。根据临床前和成人试验的数据情况,儿科的剂量确认/寻找阶段试验可以纳入富集人群或者all comer。还有一种设计思路是在剂量确认/寻找阶段纳入all comer,而在基因型或病理类型特异的剂量拓展阶段纳入富集人群,来确定药物的疗效。具体的设计应综合考虑各种证据,评估all comer有获益的可能性。如果生物学逻辑支持all comer都有潜在获益的可能性,药物没有明确的生物标志物来预测疗效,那应该纳入all comer,因为这样会加快入组速度,快速找到安全的剂量和给药计划。
但是,如果成人试验、临床前数据、科学逻辑都表明all comer获益可能性极低(比如靶向药物),应仅纳入富集人群。富集人群队列也可以在试验进行的过程中被加入,如果主队列已经入满了没位置了。为了更有效地利用数据,可以采用一些统计学方法,如模型引导的方法(持续重评估法)。在确定抗肿瘤活性时,应根据疾病或生物标志物,将研究对象限制在未来临床开发的目标人群中,或进行部分富集。可以允许先前接受过单药治疗的患者入组,因为这可以通过显示先前耐药人群的反应来揭示联合用药的益处。
确定起始剂量。利用成人推荐的2期临床剂量(Recommended Phase 2 Dose,RP2D)外推,根据体重或者BSA 调整计算得出儿童剂量后进行剂量确认,应是首选策略,而不是从头进行剂量探索。如果药物安全性隐患大、治疗指数较窄、毒性重叠,或者预期有药物间相互作用,可能需要从比成人RP2D 低20%-30%开始进行更为保守的剂量确认。剂量确认法可缩短研究时间。剂量确认队列的样本量,应能估算PK参数,并确定儿童的复方推荐剂量 (RDC)。
探索剂量和给药计划。根据EMA指南,研究设计应做到,对于观察到的不良反应,能区分其来源,以便根据不良反应情况指导药物剂量调整。例如,如果已知一种药物容易引起某种不良反应(如皮疹),则应首先降低该药物的剂量。一般来说,当新药与已明确毒性谱药物或者标准治疗联合使用时,应首先对新药进行剂量调整。此外,还应考虑基于机制的协同毒性的临床前证据。药物剂量降级或者升级的方案应该事先制定好,以避免在试验进行途中不得不暂停来修订计划。
临床前对药物的协同作用进行评估,可以为联合用药的最佳剂量提供指导。然而,为确定联合用药的最佳药物剂量和给药计划,尚需临床阶段的大量探索。剂量递增策略可包括使用交替增加每种药物剂量(或改变给药计划)的队列,或使用一种药物的标准单药剂量和给药计划,只增加新药的剂量。一般来说,如果只有两三个剂量水平,那么Rolling-6设计是可以接受的。但是,如果联合用药的剂量水平较多,部分排序 CRM(POCRM)方法(表 3)通常能更准确地确定联合用药的剂量和疗程。POCRM方法可以减少观察列表,并行地测试多种剂量和给药方案。
推荐的拓展剂量。很多时候无法用药效动力学数据来确认一个最佳的生物剂量,因为缺乏有效的生物标志物。临床前的数据和成人患者的数据可以为儿童患者用药剂量提供参考。剂量优化和剂量探索取决于毒性,耐受性和成人患者药代动力学的个体间差异。
传统上来说,将剂量爬坡阶段,第一个给药周期观察到的剂量限制毒性(DLT)的剂量,称为最大耐受剂量(MTD),但是推荐的拓展剂量不应只考虑DLT,因为它只反映了第一个给药周期的急性毒性,而应全面考虑其他的信息,比如慢性累积性毒性,后续用药过程中因毒性发生的剂量调整,PD和PK数据,以及那些对生活质量有较大影响的毒性。可以利用治疗药物监测(Therapeutic drug monitoring,TDM)来做剂量优化,如果患者吃到最大剂量、达到稳态血药浓度、耐受良好,但TDM显示暴露量偏低,提示更高剂量的药物可能带来进一步获益,这时候可以用患者内的剂量爬坡来优化患者用药。
对于精准医疗的分子靶向药,有必要在注册临床之前认真确定药物的剂量和给药计划,正如FDA的Project Optimus强调的。粗糙的剂量确认可能会损害药物的获益。由于确认药物组合中各组分最佳剂量需要较大规模的临床试验,因此应该先在成人患者中探索最佳的剂量-效应关系和暴露-效应关系,然后再外推到儿童。
在联合用药试验设计中,纳入一个单药治疗阶段。如果新药既往没有儿童数据,则应该在联合治疗之前设计一个单药给药阶段,来更好地表征该药物的剂量、PK、毒性,以及是否具有单药的疗效。单药治疗阶段起始剂量应该是100%经体表面积调整的成人剂量,除非有特殊毒性、药物相互作用、或者窄治疗窗等问题。临床试验的设计应允许患者在数个疗程后从单药治疗转入联合治疗,除非单药就观察到明显的疗效。在满足试验要求的前提下,应让尽可能少的患者暴露于单药治疗。
总结选择起始剂量和剂量探索的设计。 表3和图1呈现了早期阶段联合用药试验的潜在场景。FDA关于novel-novel combo的一份指导文件也适用于儿童适应症的开发。这份文件强调了析因分析。
在早期的药物联用试验中评估抗肿瘤活性。随机对照试验,新药+SoC vs Soc,这样的设计能最好地呈现新药在药物组合中疗效和毒性。EMA接受统计学效能不足的试验。后线交叉设计或许是可以的。相比于单臂的二期临床试验,这样的设计可以让疗效评估更稳健,减少混杂因素的影响,如患者选择,早线治疗,年龄,等等。相对较小的随机扩展阶段、随机选择或筛选设计对于疗效评价很有价值。新颖的设计(如贝叶斯设计或两阶段minimax Jung设计)可用于将样本量降到最低,每个队列 25-35 例患者,具体取决于确定样本量时使用的目标和假设。在这种情况下,控制 I 型误差(假阳性)不如控制 II 型误差(假阴性)重要,即宁可错付、不可错杀,因为此类试验的目的是确保如果一种方案更优,那么它被选中的可能性就很大。
另一种方法是在 II 期随机试验中选出优胜者。可以采取2:1随机化设计来加快入组(相比于SoC,患者可能更倾向于/更不愿意接受新疗法),但是这种设计会降低统计效能,因此只在预期药物疗效较强的时候采用。疗效判定一般选用反应率或者无进展生存期等终点,以为后续的评估提供进一步信息。如果终点事件发生率较低,可能需要全球多中心的试验来入组足够多的患者。
使用 Ensign(三阶段设计,允许进行两次中期分析)或 Simon 的两阶段设计,通过使用稳健的历史对照或已知对单药有抗药性的人群进行单臂试验来评估药物活性有一些缺点,尤其是在缺乏有意义的对照数据的情况下。
如果研究中剂量确认部分的患者接受了儿科 RDC,且疾病状态适合评估反应终点,则可将其纳入剂量拓展队列。所有应答和无应答(即使是在较低剂量水平下观察到的应答)均应报告,在某些情况下,长时间的疾病稳定也可能属于有临床意义的疗效反应。
现有的儿科联合用药早期阶段试验。表 4 列出了 ClinicalTrials.gov 数据库中纳入了儿童患者的287 项试验中,已发表的儿科联合用药早期阶段试验的实例。
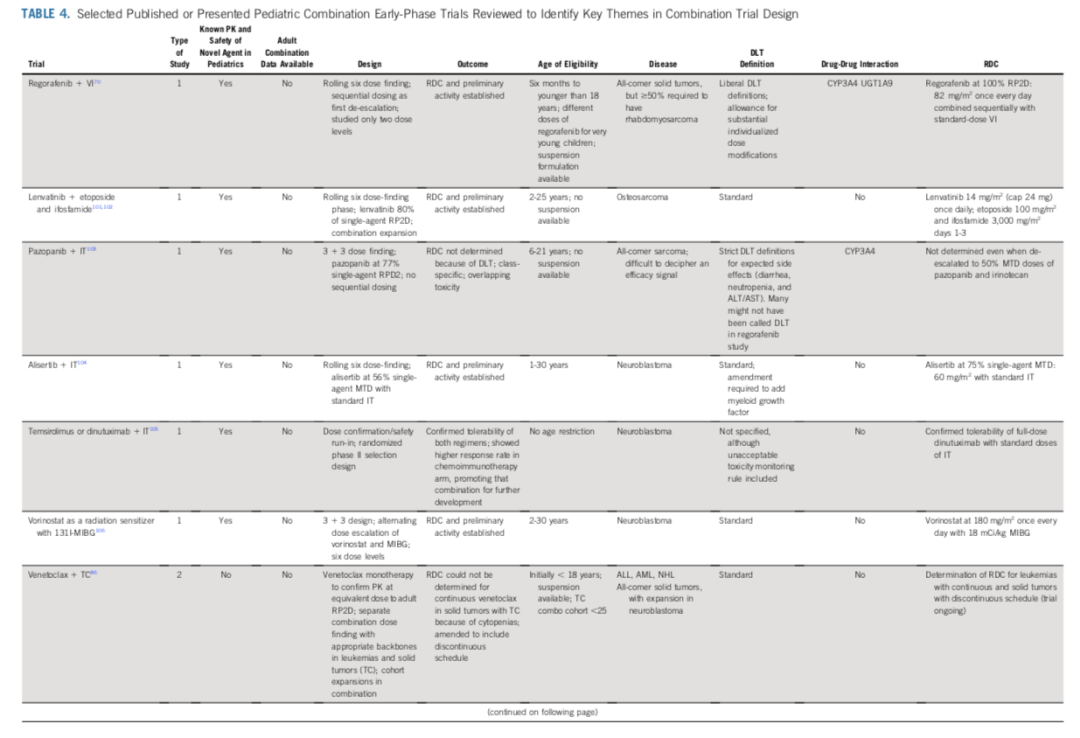
瑞戈非尼+长春新碱+伊立替康的 I 期研究是对单药瑞戈非尼试验(ITCC-047)的修正。该联合疗法采用序贯给药,DLT定义宽松,利用一项平台试验(FaR-RMS)迅速推进,进入横纹肌肉瘤的二线疗法(与标准疗法相比)。
来伐替尼+依托泊苷+伊佛酰胺的I/II期研究(ITCC-050)包括一个联合剂量探索阶段和一个针对骨肉瘤患者的联合剂量拓展阶段。在这项试验之后,OLIE(ITCC-082)随机研究评估了来伐替尼+伊佛酰胺+依托泊苷 vs SoC伊佛酰胺+依托泊苷,适应症为复发/难治性骨肉瘤)的比较。如果在初始方案中明确随机对照设计,并在I/II期部分之后进行,试验本可以进一步加速。
达拉非尼和曲美替尼联合疗法试验从单药队列开始,以确定曲美替尼的 RP2D。这导致了达拉菲尼和曲美替尼的有限剂量爬坡,随后在 BRAF V600 突变的低级别胶质瘤患者中进行联合用药剂量拓展。在儿科低级别胶质瘤中,与标准治疗(卡铂和长春新碱)相比,该联合用药的总体反应率和中位无进展生存期均更优。
总之,患有癌症的儿童和青少年应该尽早获得临床试验中的创新药物,并且需要尽快评估那些可能有益的药物。临床试验应使尽可能多的患者受益。应根据药物的作用机制、癌症生物学特性、可靠的临床前评估以及已知药物的临床活性(表 5)来开发药物组合。作者们建议,最有效的早期联合用药试验,应包括1)剂量确认/寻找;2)在目标人群中的随机化的剂量拓展队列。
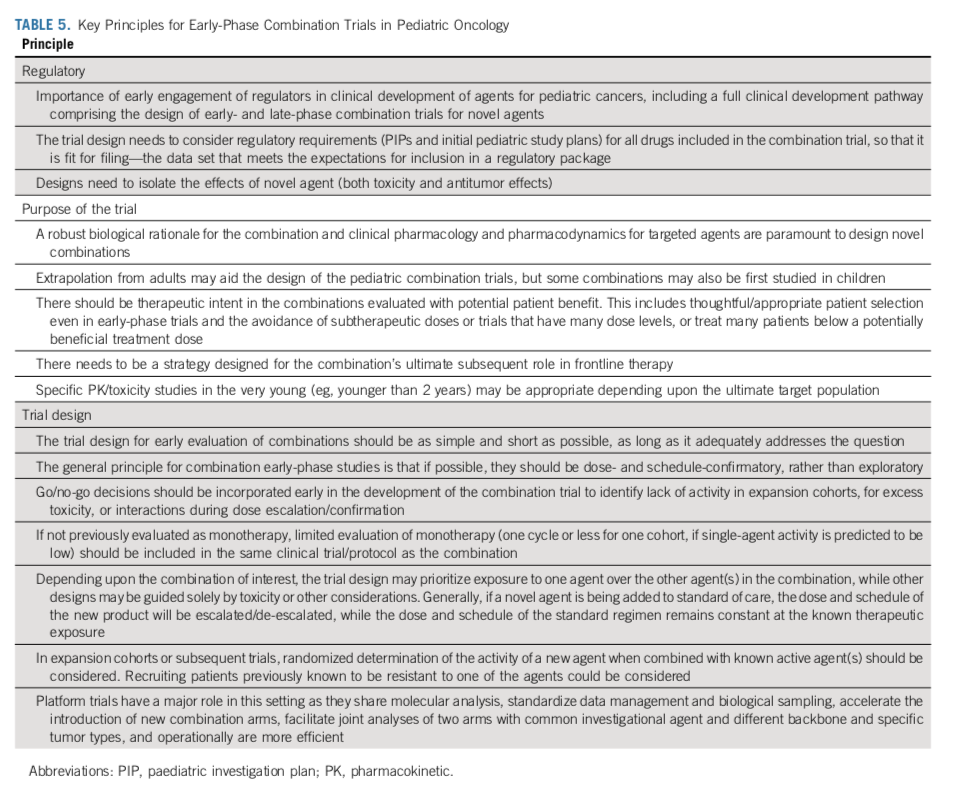
尽早与监管机构讨论试验设计也至关重要,能使试验同时达到科学和监管目的。让家长和患者倡议组织多多参与这些早期阶段的工作,有助于揭示潜在的矛盾点和错误信息,促进患者招募和患者教育,并有助于提高按时完成计划招募的可能性。此外,这种方法还能以最简便的方式对可参与临床试验的罕见病患儿进行评估。采用适合目的的方法设计早期儿科联合试验将使所有利益相关者受益,尤其是癌症儿童和青少年患者。

