【Leukemia】综述:攻击白血病克隆
时间:2024-09-09 18:06:05 热度:37.1℃ 作者:网络
白血病克隆
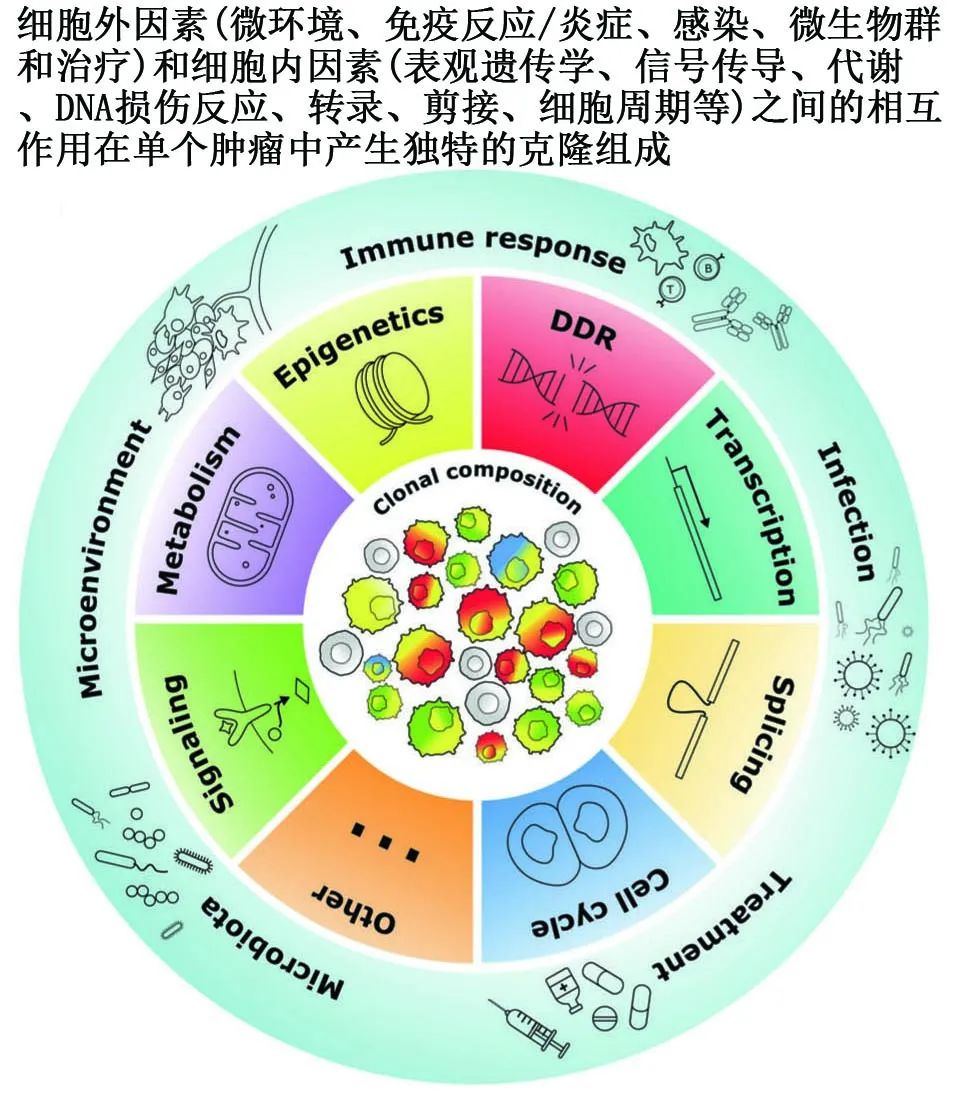
对于癌变这一现象,达尔文原理可阐明导致肿瘤内异质性的机制。癌症演化是一个肿瘤细胞适应环境以促进最佳克隆的生存和扩增的过程,该过程取决于细胞内和细胞外因素。在细胞内因素中,对能量(ATP)和构建材料(核酸、氨基酸、脂质、糖)的高需求、信号通路的失调、细胞周期的加速和对凋亡的保护产生代谢和复制应激,导致自发DNA损伤和突变积累。细胞外因素包括缺氧、应激、与微环境的相互作用、微生物群、感染、炎症和抗肿瘤免疫反应等。细胞外和细胞内的环境压力产生遗传和表观遗传改变,导致克隆选择。此外,治疗方案作为强大因素,也导致遗传学和表观遗传学畸变和克隆选择。肿瘤细胞必须适应这些细胞外和细胞内的动态挑战,从而扩大最健康的克隆。
白血病多以单克隆遗传学/表观遗传学异常作为开端,但在表现上则是一种多克隆疾病,该多克隆特性源于白血病细胞基因组/表观基因组的持续演化,以促进其生存和增殖优势。《Leukemia》近日发表综述“Star wars against leukemia: attacking the clones”,讨论了遗传学和/或表观遗传学畸变如何改变单个白血病克隆的细胞内微环境,以及细胞外微环境如何选择最适合的克隆。白血病的动态多克隆导致设计有效治疗方案极具挑战,特别是各单个白血病克隆对治疗的反应往往表现出显著差异。作者也讨论了新的治疗方法,利用单细胞多组学来识别和根除患者的所有单个克隆。

克隆演化的标志
单细胞“多组学”彻底改变了我们对肿瘤的认识,包括髓系恶性肿瘤,它是一种高度动态的多克隆疾病。遗传改变(由于持续的基因组不稳定,在微环境压力下和治疗后自发发生)、可遗传的表观遗传修饰(在微环境压力下和治疗后发生)和免疫反应之间相互作用下产生恶性克隆,并在特定条件下茁壮成长。
遗传学、表观遗传学、蛋白质组学、甲基组学和代谢组学畸变定义克隆功能异质性和协同性,它们调节克隆特异性表型,如选择性微环境优势、对免疫挑战抵抗和对治疗的敏感性。这些持续的适应性挑战是恶性克隆扩增的标志。
潜能未定的克隆性造血(CHIP)伴随着携带多种基因突变的克隆的出现,如DNMT3A、TET2、JAK2、SF3B1、ASXL1、TP53、CBL、GNB1、BCOR、U2AF1、CREBBP、CUX1、SRSF2、MLL2、SETD2、SETDB1、GNAS、PPM1D和BCORL1,并伴随着进展为血液恶性肿瘤风险的增加。CHIP与衰老、感染、化疗和/或吸烟有关,这些因素会改变组织微环境,促进含有特定突变的CHIP克隆的选择和扩增。例如,衰老有利于SF3B1、SRSF2、ATM和TET2等基因的突变;炎症应激可促进携带TET2、DNMT3A和ASXL1突变的造血克隆的扩张和进一步的恶性转化,这些突变调节DNA甲基化和染色质重塑;某些微生物群偏爱携带DNMT3A、FLT3和NPM1突变的克隆;吸烟促进ASXL1、SRSF2、SF3B1和JAK2突变的克隆。另一方面,调控DNA损伤反应的TP53和PPM1D突变克隆的扩增是由基因毒性应激和PARP抑制剂所触发。
值得注意的是,特定的克隆优势通常由两个或更多共存的突变编码。例如,当小鼠造血细胞中 Dnmt3a 或 Tet2 的联合突变改变基因表达至任一单独突变均未观察到的程度时,可产生致癌酪氨酸激酶 (OTK) 的 Flt3(ITD) 突变。此外,FLT3激酶抑制剂吉瑞替尼治疗下导致选择FLT3(ITD);NRASmut克隆,而这些突变通常在不治疗的情况下是互斥的。此外,在JAK1/2激酶抑制剂芦可替尼治疗骨髓纤维化期间,JAK2和CALR突变克隆中出现了KRAS和NRAS突变。
突变获得的顺序似乎并非随机,不分表观遗传修饰因子如DNMT3A、ASXL1、TET2或IDH1/2的突变更可能发生在早期,而FLT3(ITD)和影响细胞内信号传导的RAS基因的突变更可能发生在转化和疾病进展的后期。此外,对1540例AML患者样本的综合研究显示出一种独特的、反复发生的共突变模式,例如NRAS(G12/G13)而非NRAS(Q61)与NPM1突变共同发生,从而证实了突变(即使在同一基因内)及其组合的影响可能不同。因此,白血病的发生是一个有序和特定的演化过程,而非偶然的事件链。
肿瘤细胞及其周围微环境构成一个动态的生态系统。骨髓微环境(niche)缺氧是包括白血病在内的大多数肿瘤的标志,是肿瘤微环境的关键因素,可促进肿瘤克隆的生长、逃避免疫反应、调节其对抗肿瘤药物的反应。
遗传学和表观遗传学畸变调节对治疗的反应
关于遗传学和表观遗传学畸变如何调节对各种药物的敏感性,目前有许多报道(下表)。这些发现导致了成功的临床试验,从而改变了抗肿瘤治疗的模式。
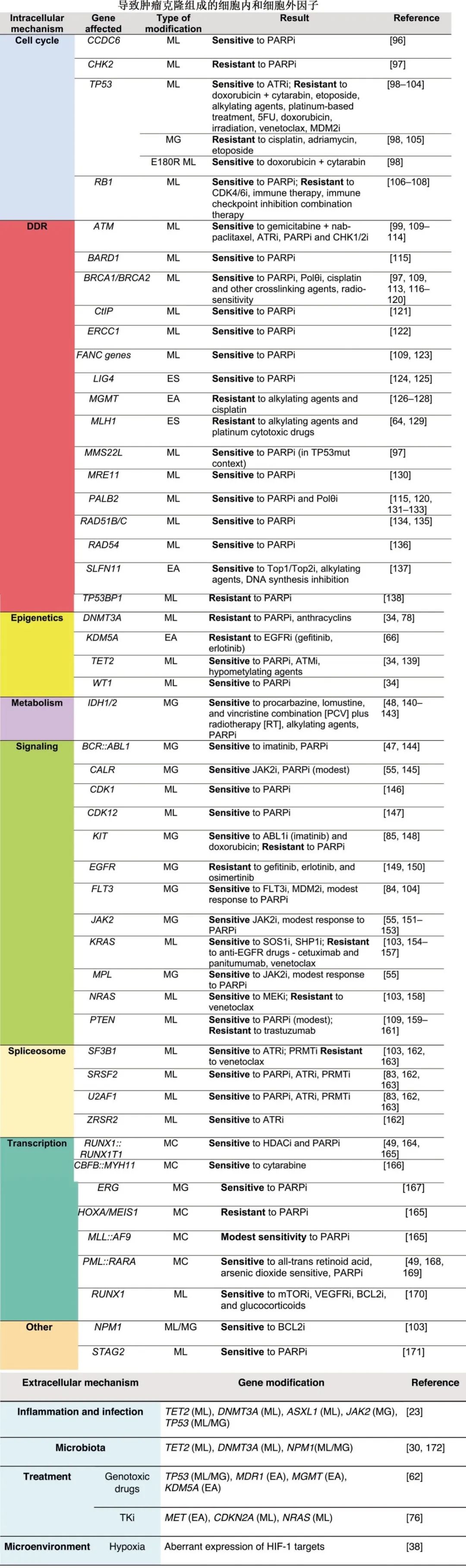
例如,由突变、易位或扩增引起的异常激活产生的OTKs与癌症的诱导、维持和恶性进展有关,因此OTKs已成为药物发现的主要靶点,并产生许多FDA批准的治疗方式。与特定治疗相关的特定癌基因的另一个例子为表达PML::RARA融合蛋白的急性早幼粒细胞白血病细胞,其对全反式维甲酸和三氧化二砷非常敏感。
探索合成致死理论(synthetic lethality)作为治疗方法,为关注与特定药物敏感性相关的癌症相关基因组和表观基因组异常的新发现铺平了道路。在这一领域,PARP抑制剂在BRCA1/ brca2突变肿瘤中引发的合成致死彻底改变了乳腺癌和卵巢癌的治疗,也改变了其他携带同源重组(homologous recombination,HR)基因突变的癌症的治疗;然而BRCA1/BRCA2基因突变仅在1-2%的成人急性髓性白血病(AML)中检测到。此外,其他DNA双链断裂(DSBs)修复基因如ATM、PRKDC、ATR、RAD51和RAD54的基因组改变在急性白血病中<1%。
尽管DSBs修复基因突变在血液恶性肿瘤中很少见,但PARP抑制剂在引发白血病的合成致死方面得到应用。例如,基因表达和突变分析(GEMA)发现一个急性白血病队列,显示BRCA1/BRCA2表达水平下调,对PARP抑制剂敏感。此外,RUNX1::RUNX1T1、PML::RARA、IGH::MYC、TC3::HLF易位和KDM6A、IDH1/2、TET2和WT1突变损害BRCA1/BRCA2依赖性HR介导的DSBs修复,从而使白血病细胞对PARP抑制剂敏感。该结果为许多已完成和正在进行的PARP抑制剂治疗血液恶性肿瘤的临床试验提供了基础。
此外,FLT3和JAK1/2激酶抑制剂可分别在表达FLT3(ITD)和JAK2(V617F) OTKs的AML和骨髓增生性肿瘤(MPN)细胞中诱导BRCA1/BRCA2缺失和对PARP抑制剂的敏感性。后一种结果导致最近开展新的临床试验,联合JAK2抑制剂pacritinib和PARP抑制剂talazoparib治疗芦可替尼难治性MPN (NCT06218628)。
除了DSB修复缺陷外,在白血病中还发现了其他具有潜在治疗应用价值的DNA损伤反应(DDR)机制的失调。例如,在一组新诊断的急性淋巴细胞白血病(ALL)患者中发现由于调节MSH2降解的基因体细胞缺失而导致的错配修复(MMR)缺陷。DNA聚合酶β抑制剂oleanolicacid 治疗后,可通过增加细胞无嘌呤/无嘧啶位点、DNA链断裂和凋亡,导致MMR缺乏ALL的合成致死。此外,碱基切除修复(BER)糖基化酶OGG1下调的AML细胞对阿糖胞苷更敏感。此外,XPD(D312N) 和 XPD(K751Q) 基因型在继发性 AML 患者中达到完全缓解的几率更高,之前与更复杂 DNA 病变(如体积较大的 DNA 加合物)的次优核苷酸切除修复 (NER) 相关。
表观遗传学是DNA和组蛋白的动态和可遗传修饰,它们独立于DNA序列调节基因活性,并可促进致癌和调节药物反应。例如,甲基化导致的MLH1和MGMT表达缺失分别导致对铂细胞毒药物和替莫唑胺的耐药。另一方面,KDM5A表达升高可引起顺铂耐药。因此,DNA甲基转移酶(DNMT)、组蛋白乙酰转移酶和去乙酰化酶(分别为HAT和HDAC)以及组蛋白甲基转移酶和去甲基化酶(HMT和HDMT)的抑制剂已开发出来并用于治疗癌症。
针对表观遗传修饰的药物如DNMT和HDAC抑制剂在白血病中的临床应用已进行探索,但成功率有限。DNMT和/或HDAC抑制剂与其他药物(如PARP抑制剂)的联合用药正在开发中,但在白血病患者的遗传学和表观遗传学畸变选择方面仍缺乏明确指南。最近对EZH1/EZH2双抑制剂valemelostat在成人T细胞白血病/淋巴瘤中的研究揭示了突变依赖性反应。例如,具有不良预后变异的恶性克隆(包括PRKCB、TP53、IRF4和PDL1)对抑制剂反应良好,而携带FN1、CCR4、MYO7B和TET2突变的恶性克隆则不太敏感/耐药。
抗肿瘤药物反应的复杂性由于共存遗传学和表观遗传学畸变而成倍增加,这些畸变可能产生与特定功能增益相关的突变协同性。此外,大多数白血病的多克隆组成导致增加了另一个挑战,因为单个克隆可能继承独特的和重叠的遗传和表观遗传畸变,这些畸变可调节单个克隆的药物反应。因此开发预测治疗反应的克隆性生物标志物至关重要。
克隆性生物标志物定义对药物的反应
基因组分析鉴定了预先存在的可追踪的敏感和耐药肿瘤克隆,并揭示了涉及药物反应的畸变。例如,携带扩增MET的克隆对EGFR激酶抑制剂吉非替尼耐药,而CDKN2A缺失的克隆对trametinib (MEK1/MEK2激酶抑制剂)和palbociclib (CDK4/6激酶抑制剂)联合使用敏感。
对于白血病,携带IDH1突变的AML克隆似乎对ABL1激酶抑制剂ponatinib更敏感,而具有NRAS突变的患者对EGFR激酶抑制剂pelitinib耐药。此外,携带DNMT3A突变的AML克隆对蒽环类药物耐药,而柔红霉素与阿糖胞苷联用是7+3方案的一部分。此外,携带RUNX1::RUNX1T1和CBFB::MYH11易位的AML细胞似乎对阿糖胞苷更敏感。SF3B1和TP53突变是慢性淋巴细胞白血病(CLL)化疗患者快速克隆演化和疾病进展的危险因素。
恶性克隆通常携带一种以上的突变,这些突变可能相互干扰。例如,虽然表达FLT3(ITD)激酶的AML克隆似乎对激酶抑制剂克唑替尼更敏感,但DNMT3A突变的共同出现预示着不仅对激酶抑制剂耐药,而且对阿西替尼(p -糖蛋白外排转运蛋白抑制剂)、cediranib (VEGFR抑制剂)、ponatinib (ABL1激酶抑制剂)和tofacitinib (JAK1/3激酶抑制剂)耐药。
AML和MPN细胞通常积累自发DNA损伤,包括由代谢产物和复制应激诱导的高毒性DSBs。AML和MPN克隆激活由克隆特异性突变调节的DDR机制,从而在内源性基因毒性应激下存活,表明DDR是一个合理的治疗靶点。例如,FLT3(ITD)、JAK2(V617F)、MPL(W515L)、CALR(del52)、TETmut、DNMT3Amut、RUNX1::RUNX1T1、RML::RARA、SRSF2mut、IDH1mut和KITmut通过调节DNA修复蛋白BRCA1和BRCA2(参与HR)和Polθ(微同源介导的末端连接的关键因子)的表达来调节DDR,影响白血病细胞对PARP、RAD52和Polθ抑制剂的反应。
这些突变也可能相互干扰以调节DSB修复。虽然携带FLT3(ITD)、NRAS、JAK2(V617)、CALRmut或MPLmut的TET2mut克隆对PARP抑制剂敏感,但携带DNMT3Amut的TET2mut克隆对PARP抑制剂耐药;值得注意的是,FLT3(ITD)、DNMT3Amut、TET2mut克隆可恢复对PARP抑制剂的敏感性。此外,RUNX1-RUNX1T1、FLT3(ITD)克隆对PARP抑制剂敏感,而RUNX1::RUNX1T1、KITmut耐药。CBFB::MYH11、KITmut和CBFB::MYH11、NRASmut也分别与PARP抑制剂的克隆性耐药和敏感性相关,但与多柔比星无关。
总的来说,上述结果突出了调节基因毒性药物、激酶抑制剂和DDR抑制剂克隆反应的遗传特征的复杂性。单细胞“组学”(如转录组学、基因组学、表观基因组学、蛋白质组学、代谢组学)和最近同时整合各种“单组学”的单细胞“多组学”,或有助于更好地理解癌症多克隆组成和治疗脆弱性。最近,对众多scRNAseq数据库的综合分析,促使产生了人工智能(AI)驱动的PERCEPTION方法,以预测多发性骨髓瘤和乳腺癌对靶向治疗的反应。
克隆攻击
个性化医疗是一种考虑患者肿瘤遗传学/表观遗传学/代谢组学/免疫特征差异来定制疾病预防和治疗的方法,而克隆医学是一种创新的方法,根据克隆独特的遗传学/表观遗传学/代谢组学/免疫组成来定制消除肿瘤克隆的治疗方法。
肿瘤演化是导致癌症异质性的原因,在制定治疗方法时必须考虑到这一点。肿瘤克隆对治疗反应的异质性早在45年前就已提出,肿瘤克隆由于其独特和重叠的“多组学”特性,对各种药物的反应不同,因此成功的治疗方案必须同时根除所有恶性克隆。
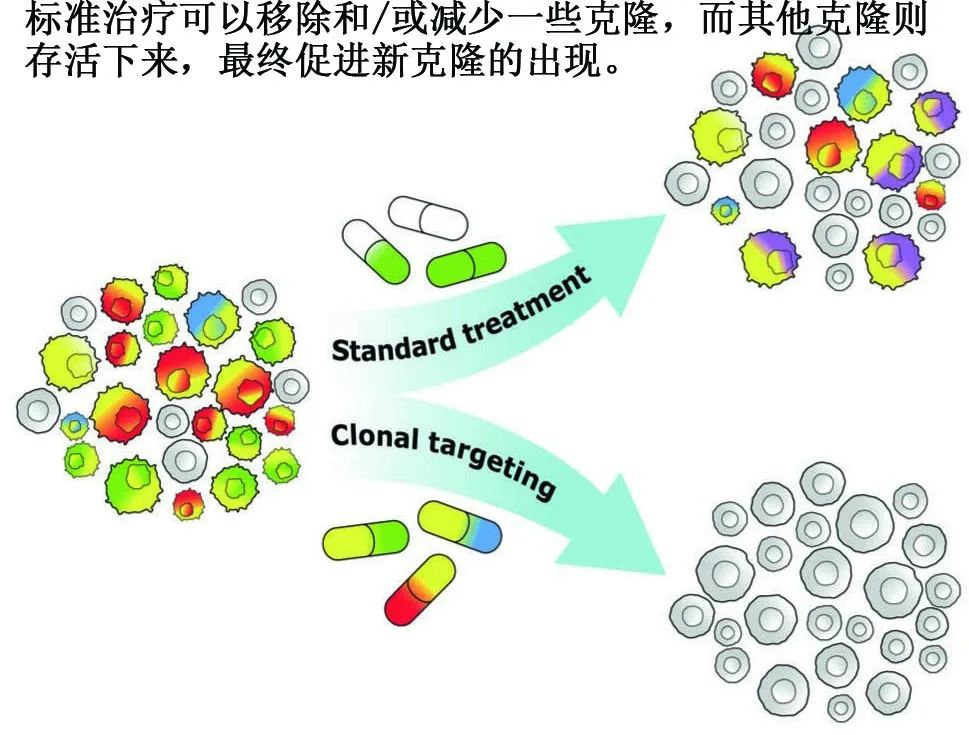
例如,AML和MPN是异质性疾病,克隆异质性阻碍了治疗的有效性。白血病样本中的克隆表现出对当前药物的反应差异,导致疾病复发,可来源于现有克隆的治疗选择和/或由于而治疗压力诱导的克隆演化。患者的恶性克隆的遗传特征可能极为复杂,因为单个克隆可以携带多个突变,这些突变对单个克隆来说是重叠的和唯一的。
最近设计了一种成功的多克隆攻击来根除由于持续代谢应激而积累DNA损伤的AML和MPN克隆。同时攻击患者样本中所有克隆的DDR抑制剂的组合可在体外和体内根除该疾病。DDR抑制剂的“克隆攻击”将基因毒性治疗的范式从使用非歧视性细胞毒性药物转变为选择性地攻击AML和MPN克隆中的DDR漏洞。FDA批准的药物(如TKI、去甲基化、基因毒性和促凋亡药物)与DDR抑制剂联合治疗的潜力还需要探索,以评估标准治疗是否能增强DDR抑制剂对抗恶性克隆的有效性。
由于克隆异质性和DNA损伤是癌症的标志,“克隆攻击”或可开启癌症治疗的新时代,并广泛适用于寻求治愈。然而,将“克隆攻击”融入日常临床实践还需要两个步骤。首先必须收集广泛的实验性单细胞“多组学”数据,为人工智能(AI)生成足够的信息,以有效地解决肿瘤克隆组成并预测个体克隆对治疗干预的敏感性。其次,医院必须有相应人员和设备,能够进行“多组”分析,并解释每天的数据。
参考文献
Monika M Toma, Tomasz Skorski .Star wars against leukemia: attacking the clones.Leukemia . 2024 Sep 2. doi: 10.1038/s41375-024-02369-6.

