述评 | 深度认识基因变异解读,提升神经遗传病的精准诊疗能力
时间:2024-10-21 18:01:24 热度:37.1℃ 作者:网络
神经遗传代谢性疾病是一大类临床诊断困难、发病率低,但种类繁多的疾病。而且临床表现多样,分子机制复杂,临床和遗传异质性极高,多数临床医生无法做出准确的临床诊断并给予精准治疗。二代测序技术(next generation sequencing,NGS)已成为临床诊断中的最重要工具之一[1]。与传统分子检测技术不同,NGS技术支持对大量基因同时进行检测,一次性获得海量的遗传数据,具有快速的大规模筛查能力。现阶段,遗传解读最重要的工具是美国临床遗传和分子病理学会(American College of Medical Genetics and the Association for Molecular Pathology, ACMG/AMP)开发的变异解读指南[2-3]及ClinGen更新建议。该指南参照各种数据库及最新报道的文献,对所有基因和位点进行基因-疾病临床证实、变异致病性、剂量敏感性等关键步骤进行解读,最终将变异分为“致病性”“可能致病性”“意义不明”“可能良性”或“良性”五个类别。
随着技术的广泛使用,变异解读问题日益凸显。通过NGS技术筛选出的大量变异,需要结合生物信息学、遗传学及功能学等分析手段,方能准确判断其致病性及与疾病的关系。目前造成该类疾病诊断困难的主要问题是:解读人员需要具有扎实的专业知识,广泛查阅文献资料,并熟练运用ACMG/AMP变异解读指南的能力。然而,在实际操作中,临床医生和检测机构均存在不足。基因检测机构虽能对检测到的变异进行生物学阐释,但普遍缺乏临床专业知识背景,难以做出与临床相符的基因诊断;而大多数临床医生缺乏遗传学知识,对于基因检测方法的选择和结果解读存在困难。
首都医科大学宣武医院神经内科一直致力于探索临床医生易于应用的遗传代谢病诊断新模式。自2018年始,我们逐步建立了一套以精细化临床表型为基础,整合生物信息学、基因功能学等多学科知识的遗传变异临床解读新路径(详见图1的“五步法”分子会诊流程)[4]。“五步法”分子会诊流程主要包括:① 分子会诊申请;② FastQ原始数据上传;③ 来自临床专家、遗传学专家和科研工作者的数据重分析;④ 由多学科团队(MDT)整合相关数据,依据临床表型、遗传数据和功能学证据进行讨论,得出相关的临床意见(明确阳性诊断、明确阴性诊断或进一步完善相关检查);⑤ 长期随访。6年期间,通过建立神经疾病多学科分子会诊门诊等形式,该MDT团队已为儿科、神经内科、神经外科、妇产科等学科提供了1000余例疑难病的临床和分子会诊,对深度认识遗传变异临床解读标准流程及其在推进临床医学精准诊疗方面的作用有不少切身体会,主要体现在如下3个方面。
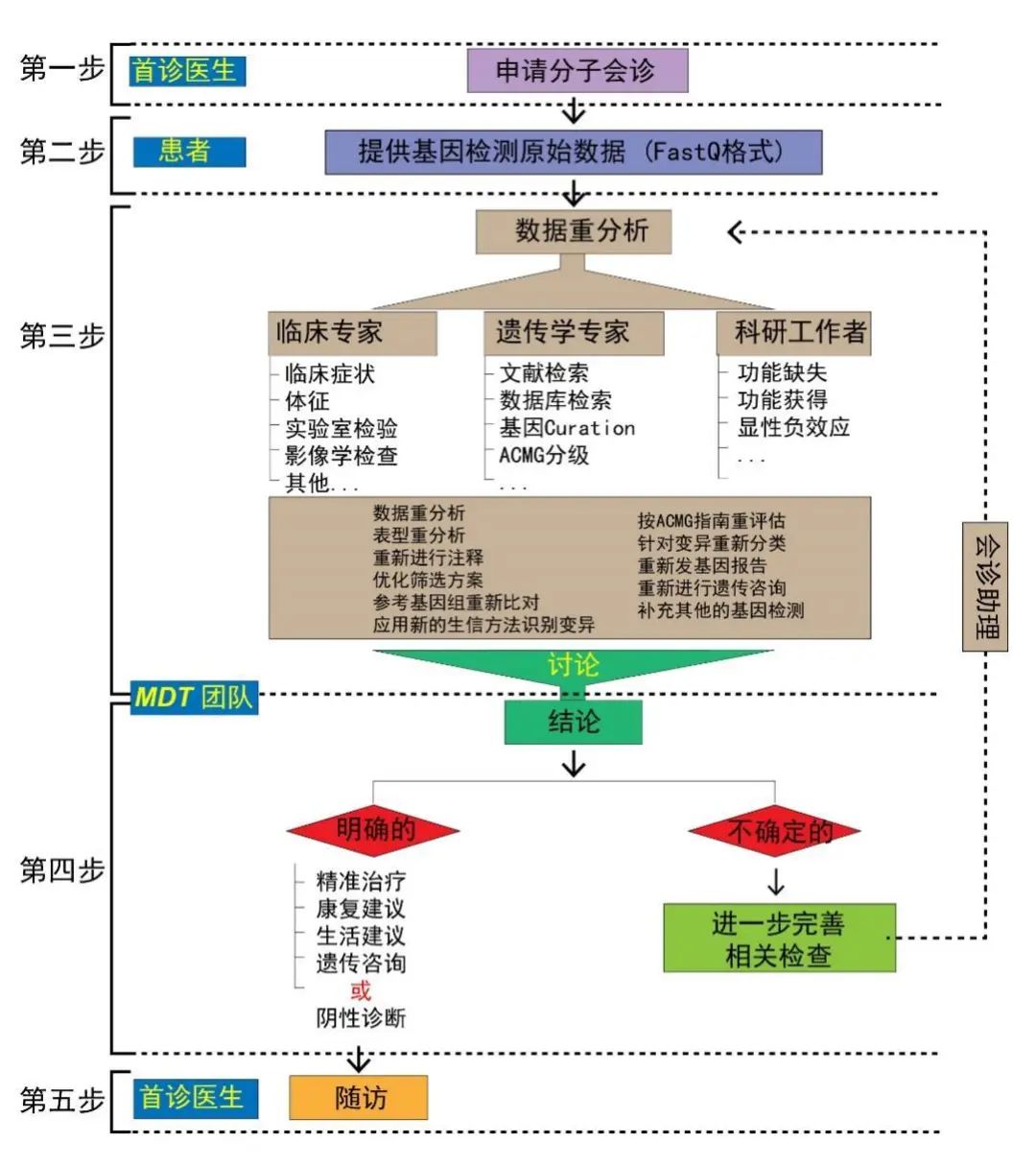
图1 “五步法”分子会诊流程
1 一线临床医生要积极参与到基因变异临床解读中来
基因变异临床解读,可以说是以“基因-变异-疾病”三者之间关系为核心,从临床表型出发(“表型驱动”的基因送检策略),最终仍落脚于临床(基因诊断及治疗)的一门学问。伴随分子生物学诊断要求的不断提高,很多复杂的基因分析和解读,需要强大的临床分析能力。临床医生在基因变异临床解读中占据着举足轻重的地位,贯穿整个检测过程。
1.1 检测前
我们应该选择哪些检测技术、送检哪些家庭成员(涉及家系的详细询问及临床证实)、如何跟患者及家属交代基因检测效力等问题。例如:很多成人相关疾病受环境修饰因素的影响较大,多重病因混杂,因此经常会存在采集不完整、特殊表型采集缺失的情况。以成人最常见的脊髓小脑性共济失调(spinocerebellar ataxia,SCA)为例,该病具有高度遗传和临床异质性。临床上往往表现为共济失调,虽然病程进展缓慢,但儿童与成人患者的临床表现有一定差异。①儿童SCA:在先天性共济失调中,共济失调的临床表现通常在1~2岁前并不明显。婴儿表现为肌张力减退伴吮吸困难及运动标志延迟,在儿童开始走路后共济失调症状渐有明显表现。检测年龄较大儿童的共济失调需评估一字步(步态共济失调)、指鼻试验与跟-膝-胫试验(辨距困难)、快速交替运动(轮替运动障碍),并关注宽基底步态及行走不稳(如扶椅行走或在支持下行走),且应关注大部分儿童伴发一定程度的学习障碍。②成人SCA:在成人共济失调中,共济失调的临床表现及严重程度与SCA类型、变异拷贝数大小均有较大关系。在缺乏家族史、长期酗酒、伴随其他小脑或脊髓病变的情况下,基因送检策略的制定及明确诊断更为困难。
1.2 检测中
临床表型特异性对变异位点解读证据有重要影响,只有临床医生可以更准确地使用。比如,PP4证据的使用指南[5]明确提出,其应符合如下条件:(a)临床检测的灵敏度高,大多数带有该基因致病突变的患者都被检测为阳性;(b)患者有某种明确综合征的症状,与其他临床表现几乎无重叠;(c)该基因通常不存在太多的良性变异;(d)家族史与疾病遗传方式一致;(e)其他致病基因排除;(f)不适用于常见的非特异性表型,如发育迟缓或心肌病。
1.3 检测后
患者及家属最关心的是基因检测后的残余风险、检测后变异的解读和临床意义、这些变异如何在家族中传递、家属同胞患病率以及后代发生疾病的风险评估与疾病治疗等。这些需由专科临床医生和遗传咨询师共同进行解答。但目前,我国遗传咨询师职业仍属空白,这些问题均由临床医生一并承担。
2 生信分析人员需要与临床充分结合
当前国内基因检测机构的生信分析团队是对检测的位点进行判读的关键人物,但其往往不与临床医生及患者进行直接沟通。前期,本MDT团队通过202例队列研究结果[4]发现,原始阳性报告中,26%为假阳性;原始阴性报告中,5%为假阴性。这一数字为我们敲响警钟,临床信息的缺少及知识体系的不完整性等,非常容易造成分析结果的不准确,而直接影响临床判断。主要原因分析如下:①对临床表型词汇理解不准确、分析策略有误,国际上针对临床表型分析的主要工具是人类表型本体库(Human Phenotype Ontology,HPO,https://hpo.jax.org/)。该项目提供了医学相关表型、疾病与表型关联性及其算法的数据集,包括遗传方式、发病年龄及按组织器官分类的临床特征、体征、实验室检查结果等。目前,多数罕见遗传病的诊断是基于HPO、Phenomizer (http://compbio.charite.de/phenomizer)和OMIM(https://www.omim.org/)数据库资源实现的。其诊断的最大问题是缺乏与临床疾病诊断指南和规范相对接的“诊断所需核心表型”。譬如,目前HPO中所提供的神经系统异常表型词汇仅1572条,远远不能概括众多神经遗传代谢疾病的表现。而由于罕见病的名称对于多数非本专业临床医师及生信分析人员来说非常生僻,最终导致使用者很难做出精确的选择,而易发生分析偏倚。②证据使用不规范,PP4证据使用不规范是最常见的(具体规则见上文)。其次,PS2、PM6等新发变异证据的升降级使用,受临床特异性的影响。PS4、PM5(已报道病例)证据使用要求报道案例的表型需与基因相关的表型一致。此外,PP1(共分离)证据的使用需明确表型的一致性等。面临如此形势,只有生信分析人员与临床充分结合,才能最大限度提高报告及诊断的准确性。
3 科研人员参与临床实践的重要价值
基础研究与临床实践脱节的现象一直存在,遗传病领域也不例外。根据基因-疾病关系评估指南[6]及ACMG/AMP变异解读指南[2]可知,基因变异临床解读是更加符合精准医疗时代循证医学的临床实践。它不仅仅依赖于临床病例的证据,实验数据也是其中不可或缺的组成部分。据报道,每年大约有250个新的基因和9200个变异被证实与疾病关联[7]。在报道病例缺乏的情况下,基础研究就是最重要、最有力的辅助手段。比如,无义变异PVS1证据在ACMG/AMP指南中,仅适用于以功能丧失机制致病的基因[8]。如果对照组设置较好,并且能重复证明待分类变异具有破坏性的影响,可考虑使用PS3功能证据。证据强度可基于实验的有效性和对基因/基因产物影响的大小进行调整[9]。
最重要的是,大部分遗传病无有效治疗方法,基础研究是我们进行基因治疗探索的重要途经。随着基因治疗技术逐渐成熟,产业化加速,不断有重磅产品获批。治疗策略包括但不局限于:①基因替换,用正常基因改变致病基因,如腺相关病毒载体(adeno-associated virus,AAV)疗法等;②基因沉默,使致病基因失活,如反义寡核苷酸(ASOs)疗法等;③基因编辑,导入新的改造基因,如CRISPR/Cas9;④基因增强,增加特定基因的表达。截至目前,基因治疗技术已成功为多种遗传性罕见病和其他慢性病患者带来新的治疗手段,近年来已为生物医药领域的创新发展提供了新动力[10-11]。
综上所述,在“健康中国2030”政策的发展战略下,为人民群众提供全方位、全周期健康服务,是时代赋予的使命,但我们也面临前所未有的困难与挑战,深度认识遗传变异临床解读标准流程,将极大地推进临床医学精准诊疗及应用。
参考文献:
1. DAVEY J W, HOHENLOHE P A, ETTER P D, et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing [J]. Nat Rev Genet, 2011, 12(7): 499-510.
2. RICHARDS S, AZIZ N, BALE S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [J]. Genet Med, 2015, 17(5): 405-424.
3. ZHANG J, YAO Y, HE H, et al. Clinical interpretation of sequence variants [J]. Curr Protoc Hum Genet, 2020, 106(1): e98.
4. ZHANG L, LI X Y, XU F, et al. Multidisciplinary molecular consultation increases the diagnosis of pediatric epileptic encephalopathy and neurodevelopmental disorders [J]. Mol Genet Genomic Med, 2023, 11(11): e2243.
5. BIESECKER L G, BYRNE A B, HARRISON S M, et al. ClinGen guidance for use of the PP1/BS4 co-segregation and PP4 phenotype specificity criteria for sequence variant pathogenicity classification [J]. Am J Hum Genet, 2024, 111(1): 24-38.
6. STRANDE N T, RIGGS E R, BUCHANAN A H, et al. Evaluating the clinical validity of gene-disease associations: An evidence-based framework developed by the clinical genome Resource [J]. Am J Hum Genet, 2017, 100(6): 895-906.
7. WENGER A M, GUTURU H, BERNSTEIN J A, et al. Systematic reanalysis of clinical exome data yields additional diagnoses: Implications for providers [J]. Genet Med, 2017, 19(2): 209-214.
8. ABOU TAYOUN A N, PESARAN T, DISTEFANO M T, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion [J]. Hum Mutat, 2018, 39(11): 1517-1524.
9. BRNICH S E, ABOU TAYOUN A N, COUCH F J, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework [J]. Genome Med, 2019, 12(1): 3.
10. SCHAMBACH A, BUCHHOLZ C J, TORRES-RUIZ R, et al. A new age of precision gene therapy [J]. Lancet, 2024, 403(10426): 568-582.
11. ZHANG Y, WU Z Y. Gene therapy for monogenic disorders: challenges, strategies, and perspectives [J]. J Genet Genomics, 2024, 51(2): 133-143.
【引用格式】王朝东,李旭颖.深度认识基因变异解读,提升神经遗传病的精准诊疗能力[J]. 中国神经精神疾病杂志,2024,50(6):321-324.
DOI:10.3969/j.issn.1002-0152.2024.06.001

