FGFR靶向治疗的临床进展
时间:2024-06-13 21:00:34 热度:37.1℃ 作者:网络
前言
成纤维细胞生长因子(FGF)通过FGF受体(FGFR1-4)发出信号,协调胎儿发育,有助于组织和全身稳态,但FGFR基因的融合、重排或突变也可能促进肿瘤发生。FGFR融合可分为I型(非受体型,由融合伴侣的N端置换引起,如BCR-FGFR1和ZYM2-FGFR1)、IIa型(受体型,也由融合伴侣N端置换产生;如FN1-FGFR1、KLK2-FGFR2)或IIb型(受体型,由融合伴侣C端置换产生;如FGFR2-BICC1、FGFR3-TACC3)。
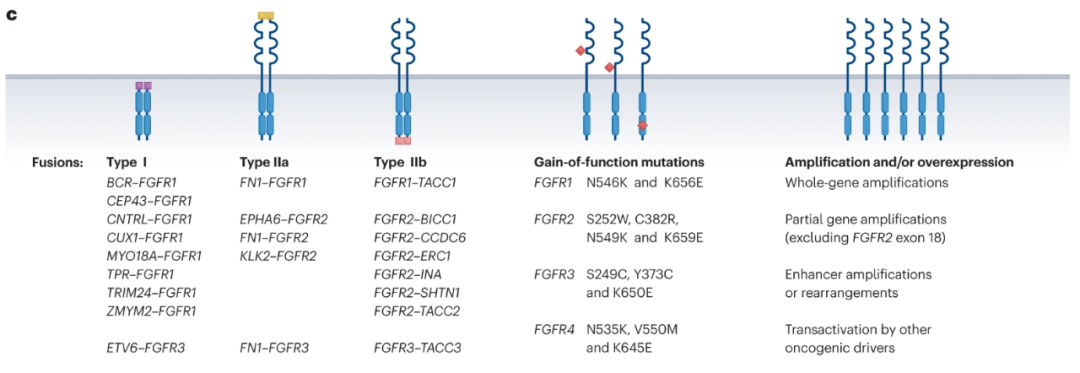
在实体瘤中检测到的FGFR变化包括非小细胞肺癌中的FGFR1扩增(20%)、乳腺癌中的FGFR 1/2扩增(7-23%)、尿路上皮癌中的FGFR3突变(10-60%)或FGFR3融合(6%)、肝内胆管癌中的FGFR2融合(10-20%)、子宫内膜癌中的FGFR2突变(12%)和胃癌中FGFR2的扩增(5-10%)。在约7.0%的未经选择的癌症患者中可以检测到FGFR基因的改变。
目前,已经开发出了多种FGFR靶向药物,包括泛FGFR抑制剂(erdafitinib和futibatinib)、FGFR1/2/3抑制剂(infigratinib和pemigatinib),以及一系列更具特异性的药物,其中一些已进入临床使用。Erdafitinib已被批准用于携带FGFR2/3突变的尿路上皮癌患者,futibatinib和pemigatinib被批准治疗携带FGFR2融合或重排的胆管癌患者。这些药物的临床益处在一定程度上受到高磷血症的限制,这是由于FGFR1的脱靶抑制以及FGFR基因中耐药性突变的出现。下一代小分子抑制剂,如lirafugratinib和LOXO-435,以及FGFR2特异性抗体bemarituzumab,有望降低高磷血症的风险,并有能力克服某些耐药性突变。
FGFR的结构和信号通路
FGFs由具有球状或非典型β三叶结构的保守核心结构域组成,可分为典型FGFs(FGF1-10、FGF16-18、FGF20和FGF22)、内分泌型FGF(FGF19/FGF15、FGF21和FGF23)以及FGF同源因子(FGF11-14)。全长FGF受体(FGFR1-4)是I型跨膜蛋白,具有三个细胞外免疫球蛋白样结构域和一个胞内酪氨酸激酶结构域。典型的FGF与硫酸乙酰肝素辅因子一起通过FGFR传递信号,而内分泌型FGF与klotho共受体(KLA和KLB)和硫酸乙酰肝素辅助因子一起通过FGFR传递信号。FGF–FGFR信号传导参与细胞存活、增殖、代谢、迁移和分化,包括生理作用,如协调胎儿发育和维持组织和/或全身稳态。这些受体也可以驱动肿瘤的发展。
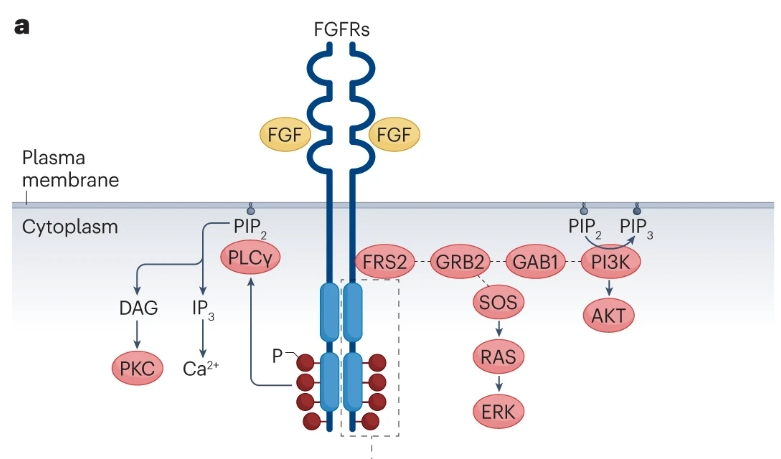
FGFR中配体依赖性二聚化或病理改变通过自身抑制机制释放而导致内源性激酶激活。FGFR激活导致FRS2依赖性激活PI3K–AKT、RAS–ERK和磷脂酶Cγ(PLCγ)依赖性二酰基甘油(DAG)–PKC和IP3–Ca2+信号。
癌症患者的FGFR改变通过异常的FGFR信号传导促进肿瘤发生:FGFR扩增导致FGFR过度表达和下游信号通路过度激活;FGFR融合或重排导致融合伴侣介导的FGFR二聚化、磷酸化改变和异常的下游信号传导;细胞外区或跨膜结构域中的FGFR突变通过改变对FGF配体的特异性或亲和力,以及配体非依赖性二聚化来激活FGFR;酪氨酸激酶结构域中的FGFR突变由于自身抑制机制的破坏而结构性性地激活FGFR。
小分子FGFR抑制剂
几种小分子FGFR抑制剂已被开发用于治疗具有FGFR改变的癌症患者,如尿路上皮癌、乳腺癌、胃癌、肝癌、肺癌、卵巢癌和宫颈癌。能够与酪氨酸激酶结构域的ATP结合位点结合的FGFR抑制剂通常分为多激酶FGFR抑制剂或选择性FGFR抑制剂。Derazatinib、dovitinib、tasurgratinib、lenvatinib、lucitanib、nintedanib和ponatinib都是多激酶FGFR抑制剂,对FGFR以外的多种RTK具有活性。然而,由于其广泛的活性,与选择性FGFR抑制剂相比,多激酶FGFR抑制剂的作用机制和不良反应都很复杂。
选择性FGFR抑制剂包括泛FGFR、FGFR1/2/3和FGFR2/3抑制剂以及选择性FGFR2、FGFR3或FGFR4抑制剂。泛FGFR抑制剂包括 erdafitinib, futibatinib, rogaratinib和resigratinib;FGFR1/2/3抑制剂包括pemigatinib, infigratinib, fexagratinib和zoligratinib;Lirafugratinib是一种选择性FGFR2抑制剂,LOXO-435是一种选择FGFR3抑制剂,roblitinib和fissotioninib是选择性FGFR4抑制剂。
目前,根据在II期试验中观察到的疗效和耐受性,美国食品药品监督管理局(FDA)在这些适应症的二线或更后线的治疗中加速批准erdafitinib、futibatinib、infigratinib和pemiginib。在erdafitinib的临床研究(BLC2001)中,根据FGFR3或FGFR2/FGFR3融合物中至少一种突变的存在,选择局部晚期和/或不可切除的尿路上皮癌患者。整体队列的ORR为40%,中位无进展生存期(mPFS)和中位总生存期(mOS)分别为5.5个月和13.8个月。46%的患者出现≥3级不良事件,在初步分析中包括低钠血症(11%)、口腔炎(10%)和乏力(7%),以及长期随访的指甲(15%)、口炎(14%)和皮肤事件(8%)。
在FIGHT-203的II期研究中,在携带I型FGFR1融合(如ZMYM2–FGFR1或BCR–FGFR2)的MLN患者中测试了pemiginib的药效和安全性。其中77%(24/31)的患者对pemiginib有完全响应。常见的≥3级不良事件包括贫血(18%)、口腔炎和四肢疼痛(均为12%)。2022年8月,FDA批准了pemiginib用于携带FGFR1融合的复发性和/或难治性MLN患者。
任何级别的高磷血症都发生在大多数接受泛FGFR抑制剂或FGFR1/2/3抑制剂的患者中。这种不良反应很可能反映了泛FGFR或FGFR1/2/3抑制剂对FGF23信号传导的抑制,因为这种分泌蛋白依赖于与近端小管上皮细胞膜上的FGFR1和klotho的结合。该复合物通过抑制磷酸钠协同转运蛋白(NPT2a/c)调节肾脏中的磷酸盐吸收。FGFR1在这一过程中的作用得到证实,因此,开发FGFR2特异性和FGFR3特异性抑制剂有望避免或降低这种不良反应的发生率。
FGFR抑制性单克隆抗体
在靶向FGF/FGFR信号的生物制剂中,bemarituzumab是目前正在III期试验中评估的唯一药物。bemarituzumab是一种人源化抗FGFR2b抗体,其抑制FGF7、FGF10和FGF22的结合,从而抑制癌细胞中配体诱导的FGFR2b信号传导,并且由于修饰的Fc结构域对自然杀伤细胞上的人FcγRIIIA受体的亲和力增加,因此具有诱导抗体依赖性细胞介导的细胞毒性(ADCC)的能力。
在一项涉及晚期FGFR2b过表达胃和胃食管交界腺癌(GEA)患者的I期试验中,bemarituzumab单药治疗显示出可接受的耐受性,ORR为18%(5/28)。随后,在携带FGFR2扩增或FGFR2过表达的GEA患者的II期FIGHT试验中,将bemarituzumab与mFOLFOX6(包括氟嘧啶、亚叶酸和奥沙利铂)联合测试。bemarituzumab联合化疗组和安慰剂联合化疗组患者的ORR分别为53%和40%,mPFS分别为9.5个月和7.4个月(P=0.073)。bemarituzumab的其他几项试验,包括评估bemarituzumab联合mFOLFOX6加(NCT0511626) 或不加(NCT050252801) nivolumab在先前未经治疗的晚期GEA患者中的研究正在进行中。
新型FGFR靶向疗法
蛋白水解靶向嵌合体(PROTAC)、嵌合抗原受体(CAR)T细胞、抗体偶联药物(ADC)、放射免疫偶联物(RICs)和可溶性受体是能够靶向FGF–FGFR信号级联的潜在新治疗模式,其中一些已经或正在临床试验中进行测试。
PROTAC
PROTAC具有双重部分,使其能够与靶蛋白和E3泛素连接酶相互作用,从而诱导靶蛋白的泛素化介导的蛋白酶体降解。LC-MB12是一种基于infigratinib的PROTAC,能够优先降解FGFR2,并抑制表达TEL–FGFR2融合的Ba/F3细胞和FGFR2扩增的SNU16癌细胞的增殖。相比之下,DGY-09-192是另一种基于 infigratinib的PROTAC,旨在募集von Hippel–Lindau蛋白(VHL),从而优先降解FGFR1和FGFR2。这些药物目前仍处于临床前开发阶段。
CAR-T细胞
由于PAX3–FOXO1的下游作用,FGFR4在儿科横纹肌肉瘤中普遍过表达。然而,N535K和V550L等耐药性突变通常在治疗前出现,这可能会限制小分子抑制剂的有效性。因此,研究人员开发了靶向FGFR4的CAR-T细胞,如RJ154-HL和3A11,具有不同的靶向FGFR4的scFv。目前,靶向FGFR的CAR-T细胞也仍处于临床前开发阶段。
基于抗体的药物
Aprutumab是一种全人源抗FGFR2抗体,它识别FGFR2b和FGFR2c亚型共同N末端区域P23、L27和E29周围的表位,并已被证明能诱导这些变体的内化和运输至溶酶体降解。Aprutumab和bemarituzumab在I期临床试验中均显示出良好的安全性和耐受性。目前,bemarituzumab已作为联合疗法进行II期和III期临床试验,而Aprutumab用于开发ADCs和RICs。
Aprutumab ixadotin(前身为BAY-1187982)是具有不可裂解连接子的抗FGFR2 ADC,LY3076226是具有可裂解连接子的抗FGFR3 ADC。然而,测试这两种药物的第一阶段试验(nct22368951)由于耐受性差和活性有限而终止。
此外,Aprutumab和抗FGFR3抗体vofatamab分别用于FGFR2靶向的RIC(227Th-Aprutumab,BAY 2304058)和FGFR3靶向的RIC,225Ac-vofatamab(FPI-1966)和111In vofatamab(FPI-1967)。BAY 2304058仍处于临床前开发阶段,而FPI-1966(用于放射免疫疗法)和FPI-1967(用于放射成像)目前正在I/II期试验(NCT05363605)中进行测试,用于表达FGFR3的晚期实体瘤患者。
可溶性FGFR受体
可溶性FGFR受体,如衍生自FGFR1c的细胞外结构域的FP-1039和衍生自FGFR 3c的细胞外区域的Recifecept,是另一类通过充当诱饵受体来捕获FGFR配体的重组蛋白药物。FP-1039已显示对促有丝分裂FGF配体具有高亲和力,并且在FGFR1扩增的肺癌和FGFR2突变的子宫内膜癌的小鼠异种移植模型中具有抗肿瘤作用。FP-1039在一期试验中具有可接受的耐受性。测试FP-1039与各种化疗联合用药的Ib期试验(NCT01868022) 显示同时接受紫杉醇和卡铂治疗的鳞状非小细胞肺癌患者的ORR为47%,同时接受培美曲塞和顺铂治疗的恶性胸膜间皮瘤患者的ORR为39%。
小结
目前,小分子FGFR抑制剂和FGFR靶向抗体药物已在临床后期试验中进行开发和测试,其中几种小分子FGFR抑制剂已被批准用于FGFR改变的癌症患者。尽管如此,某些不良事件,如FGFR1脱靶抑制引起的高磷血症和获得性耐药性,仍然限制了这些药物的临床益处。选择性第三代抑制剂,如特异性靶向FGFR2或FGFR3的抑制剂,正在临床开发中,这些药物可能避免或显著降低高磷血症的风险,并可能克服突变介导的耐药性。
参考文献:
1.FGFR-targeted therapeutics: clinical activity, mechanisms of resistance and new directions. Nat Rev Clin Oncol.2024 Feb 29

