脑胶质瘤会遗传吗?哪些综合征容易遗传后代?
时间:2023-01-12 15:02:40 热度:37.1℃ 作者:网络
最近我们收到一特别患者的疑问,“我查过很多相关资料,众说纷纭,但教科书上写的是与遗传无关,但这个病至今为止原因也还没搞透啊……患者刚做完手术,病理结果为三级,而妻子就发现自己怀孕了,现在无法抉择这孩子能不能要呢?是否有这种案例呢?比如直系亲属得了之后,而后代也发现有此病的!?”我们的医学专家团队在详细分析评估了患者情况后,给出了意见“不是遗传病,孩子遗传概率极其低” 那目前这方面研究进展如何呢?胶质瘤到底有没有可能遗传后代呢?哪些特殊情况会遗传呢?

绝大多数成人脑肿瘤的诱因尚不明确,虽然胶质瘤的发生和部分基因有关,但大部分胶质瘤不具有遗传倾向,然而研究发现有一些罕见的遗传性综合征或许会增加一个人患脑肿瘤的风险。目前研究显示,大约1%-5%的脑肿瘤由遗传综合征导致,这类遗传综合征可使神经系统肿瘤的风险增加。其中部分肿瘤与神经纤维瘤病和几种其他遗传性综合征相关,这些综合征包括1型神经纤维瘤病(NF1)、2型神经纤维瘤病(NF2)、von Hippel-Lindau综合征、Li-Fraumeni综合征(LFS)、家族性腺瘤性息肉病(FAP)和基底细胞痣综合征。
1型神经纤维瘤病NF1
1型神经纤维瘤病(neurofibromatosis, NF1)的发生率为1/3000,与17号染色体上的基因有关。NF1基因编码神经纤维瘤蛋白,该蛋白质可通过激活Ras蛋白的GTP水解作用而限制细胞增殖。可出现多发性神经纤维瘤,某些可恶变为神经纤维肉瘤。
高达5%-10%的NF1患者可出现其他恶性肿瘤,包括其他恶性神经鞘膜肿瘤(如恶性神经鞘瘤)和星形细胞瘤。星形细胞瘤通常为低级别,常具有毛细胞型组织学表现。这些病变好发于视神经通路、下丘脑和小脑。

目前有人提出,NF1中的恶性变性反映了二次打击假说:即一个等位基因在生殖系中呈组成性失活,而另一个等位基因发生体细胞失活(第二次打击)。动物模型的结果与该假说一致,但表明第二次打击可以是p53基因中的突变。
2型神经纤维瘤病
2型神经纤维瘤病(neurofibromatosis type 2, NF2)是一种常染色体显性疾病,使患者易发生多种肿瘤性病变。该病由NF2基因突变导致,这种基因是22号染色体上的肿瘤抑制基因,可编码一种叫做merlin或schwannomin的膜细胞骨架蛋白,而这种蛋白会参与肌动蛋白-细胞骨架的组成。其他修饰基因也可能与NF2有关。
具有诊断意义的发现是双侧前庭神经鞘瘤(听神经瘤),前庭神经鞘瘤可见于90%-95%的NF2患者,通常在30岁前发生。
也可见其他类型的脑肿瘤,其中最常见的是脑膜瘤。大约半数的NF2患者有脑膜瘤,且常存在多发性脑膜瘤。脑膜瘤的发病率随年龄的增长而增加,终生风险可能高达75。与散发性脑膜瘤患者相比,NF2患者往往在更小的年龄发生脑膜瘤,且脑膜瘤更常呈非典型性或间变性。
Von Hippel-Lindau综合征
von Hippel-Lindau综合征是一种常染色体显性疾病,与血管母细胞瘤、胰腺囊肿和神经内分泌肿瘤、肾肿瘤和嗜铬细胞瘤有关。该基因位于染色体3p25,在正常情况下是肿瘤抑制基因。

Li-Fraumeni综合征
Li-Fraumeni综合征(Li-Fraumeni syndrome, LFS)是常染色体显性遗传病,通常与肿瘤蛋白53(tumor protein 53, TP53)基因种系突变相关。
LFS的主要特征是在45岁前发生肉瘤、乳腺癌、白血病、肾上腺皮质癌和脑肿瘤。多种脑肿瘤可见于LFS,最常见的是婴儿中的脉络丛肿瘤,儿童中的髓母细胞瘤,以及年轻成人中的星形细胞瘤。尤其需要注意,即使没有其他癌症或阳性家族史,很大一部分脉络丛癌也与TP53种系突变相关。
除LFS标准包含的脉络丛癌之外,还未明确对脑肿瘤患者进行TP53种系检测的推荐。一些专家提出,存在其他几种脑肿瘤时,应考虑进行种系检测,因为研究结果表明,在特定组织学和分子亚组中,种系TP53突变的发生率很高,这独立于其他肿瘤的个人史和家族史。这些包括:
●免疫组化显示肿瘤存在突变型p53的儿童较低级别脉络丛肿瘤。
●儿童音猬因子型髓母细胞瘤–大规模测序工作发现,20%(13/63)的5-16岁儿童音猬因子(sonic hedgehog, SHH)型髓母细胞瘤以及8%(13/170)的所有儿童SHH型髓母细胞瘤存在种系TP53突变[117]。对儿童SHH型髓母细胞瘤应考虑进行基因检测(表 1)。
●伴罕见异柠檬酸脱氢酶1(IDH1) R132C突变的星形细胞瘤–在LFS家族中发现种系TP53突变与罕见的异柠檬酸脱氢酶1(isocitrate dehydrogenase type 1, IDH1)突变(R132C)密切相关[118],在癌症基因组图谱(The Cancer Genome Atlas, TCGA)数据库中发现2例兼有种系TP53突变和IDH1 R132C突变的患者,这表明可以推荐存在此突变的患者进行种系TP53检测。
家族性腺瘤性息肉病
家族性腺瘤性息肉病(familial adenomatous polyposis, FAP)是常染色体显性遗传病,由5号染色体上的腺瘤性结肠息肉病基因突变导致。大多数FAP相关脑肿瘤为髓母细胞瘤,但也报道过胶质瘤。
错配修复缺陷
遗传性非息肉病性结直肠癌(hereditary nonpolyposis colorectal cancer, HNPCC)的特征是DNA核苷酸错配修复相关的某种酶基因发生单个种系突变。这些患者易发生高级别胶质瘤和其他实体瘤。
MLH1、MSH2、MSH6和PMS2等错配修复基因的双等位基因突变可导致组成性错配修复缺陷(constitutional mismatch repair-deficiency, CMMR-D)综合征。CMMR-D患者在儿童期晚期可能发生脑肿瘤(主要是胶质母细胞瘤),并且可能具有NF1样表型,表现为咖啡牛奶斑和腋下雀斑。
CMMR-D相关的胶质母细胞瘤和其他肿瘤比散发性肿瘤的突变负荷要高,这预示着免疫检查点抑制剂治疗可能有效,并且至少有一份病例报告称,2例CMMR-D相关的复发性胶质母细胞瘤患儿使用纳武利尤单抗效果良好。
基底细胞痣综合征 — 基底细胞痣综合征又称Gorlin综合征或痣样基底细胞癌综合征,这类患者发生髓母细胞瘤的风险增加。该综合征由patched 1(PTCH1)基因种系突变导致,该基因是一种肿瘤抑制基因。
家族性胶质瘤
上述遗传性综合征仅解释了一部分家族聚集性胶质瘤病例,而在没有任一种已被识别的遗传综合征的家族中,关于导致其对胶质瘤明显易感的潜在遗传学因素我们仍知之甚少。共同的环境暴露也可能导致了某些家族的风险。
一项瑞典的人群研究纳入了在8年间确诊的297例星形细胞瘤患者的2141例一级亲属,发现5%存在胶质瘤的家族性聚集,分离分析纳入了14个家族,每个家族有至少2例受累一级亲属,结果显示65%(9个家族)存在常染色体隐性遗传模式,而仅21%(3个家族)疑似为常染色体显性遗传模式。
一项多国研究纳入了376个家族(每个家族至少有2例确诊的胶质瘤患者),多数家族(83%)仅有2例胶质瘤患者,其中57%为一级亲属,32%为二级亲属。家族性病例的年龄、性别和级别分布类似于文献中的散发性病例。大量家族中仅有连续两代的2例胶质瘤病例,提示这是一种外显率很低的常染色体显性遗传模式,但确切的遗传模式尚不确定。
大范围的基因测序工作可能会在某些这些家族中识别出病因性的生殖系突变。例如,一项研究对55个胶质瘤家族中的90例患者进行了全外显子组测序,发现3个主要为少突胶质细胞瘤的家族存在改变蛋白质的端粒保护1(POT1)基因变异。POT1编码shelterin复合体的一种成分,该复合体参与端粒维持并可对DNA损伤做出反应。两个较大的家族中证实了存在不完全外显,大约一半已知携带突变基因的个体发生了胶质瘤。这些家族中的其他癌症包括肺癌(n=3)、白血病(1例)、结肠癌(1例)和肾癌(1例)。家族性黑素瘤中也报道过罕见的POT1功能丧失性突变。
遗传易感性
数项全基因组关联研究(genome-wide association study, GWAS)评估了脑肿瘤风险,并识别出了与胶质瘤相关的遗传多态性,包括TERT、CCDC26、CDKN2A/CDKN2B、RTEL1、PHLDB1和EGFR。一篇大型meta分析识别出了13个新的胶质瘤易感位点,虽然某些易感位点是所有胶质瘤亚型所共有(如,TP53),但另一些位点则仅见于胶质母细胞瘤(如,EGFR)或非胶质母细胞瘤(如,PHLDB1)胶质瘤。已识别出的脑膜瘤易感位点有2个,即10q12.31和11p15.5。
采用候选基因方法也识别出了值得关注的遗传多态性。例如,涉及致癌物代谢、DNA修复、炎症/变态反应和其他免疫应答的基因变异可能会增加脑肿瘤风险。总体而言,虽然这些基因变异还不具备足够强的关联性来筛查高危人群,但这为认识致癌途径开辟了新思路。
INC国际教授疑难位置脑瘤案例解读
脑干-中脑顶盖胶质瘤伴严重脑室扩张积水
德国术后1年半考入名校、体育满分
患者情况:2020年11月,14岁的小永因患脑干-中脑顶盖部位的毛细胞型星形细胞瘤住进医院,MRI影像检查发现松果体区占位伴幕上脑室扩张积水,病灶有明显强化生长迹象。2年前父母带他去过国内多家知名医院,可是得到的治疗意见都是:“手术风险太大,无症状时不必着急手术。”父母因此一直没敢给孩子选择手术治疗,直到孩子的病情越来越严重,无法再保守治疗……
治疗过程:小永父亲在医生朋友的推荐下联系到INC德国巴特朗菲教授进行国际诊疗咨询。巴教授表示,小永已有很明显的手术指征,如果能及时接受手术,脑脊液通道可以通过肿瘤的切除而得到恢复。教授凭以往的手术经验判断,可实现神经外科显微镜下手术近全切瘤,且手术风险很低。收到巴教授的诊疗意见回复后,小永父母决定带孩子赴德国接受巴教授的手术治疗。由于肿瘤基底部和脑干关系密切、紧邻小脑,术中教授在保全患者正常功能神经和脑干等重要脑组织的前提下,完成肿瘤近全切手术,术后无新发永久性后遗症。
术后1年半:INC巴教授及国际医学顾问再次回访小永时,小永父母表示孩子手术后恢复非常好,不仅已经回归正常生活和学习,身高还从原来的一米七几长到了一米八多。今年7月,小永凭借优秀的中考成绩,考入国内知名高中,其中体育成绩还获得了满分,完全看不出来他1年半以前做过开颅手术。INC巴教授在邮件随访中表示,肿瘤至少得到了99%的切除,病灶组织病理学为毛细胞型星形细胞瘤,复发的可能性非常低。
国内知名高中校长为小永颁发录取通知书
巨大视神经胶质瘤
手术后复诊终于看清了巴教授的样子
患者情况:2018年7月,年仅5岁的小冬患巨大视神经胶质瘤,肿瘤严重压迫视神经等重要功能神经和脑组织,眼睛几乎没有光感,父母为他访遍国内知名神经外科医院,均表示这个位置无法手术,或者手术风险极大但是切除率很低,不建议治疗。看着孩子病情一天天恶化,家长心急如焚,经过多方打探才找到INC神经外科医生集团,并在INC团队的帮助下联系到世界颅底手术大师巴特朗菲教授进行国际远程诊疗。巴教授详细诊断小冬病情后表示,尽管视神经被肿瘤浸润包裹,但还是有很大机会切除肿瘤,且手术不会导致失明等症状。父母得到这样的诊疗回复,立即决定带小冬赴德国找巴教授手术。
治疗过程:巴教授根据肿瘤具体位置、大小和形态制定个体化手术方案,通过额骨颅骨开颅术和大脑半球间穹窿入路,在神经电生理监测护航下实现近全切肿瘤,术中无新发功能神经损伤,术后无新发永久性并发症。术后两周出院,小冬视力明显好转,可自行站立、短距离行走。
胶质瘤压迫几乎失明,赴德手术全切保留视力
术后1年随访,原本已经失明的小冬视力渐渐恢复了
术后3年半随访,肿瘤全切并无复发,视力较前明显好转
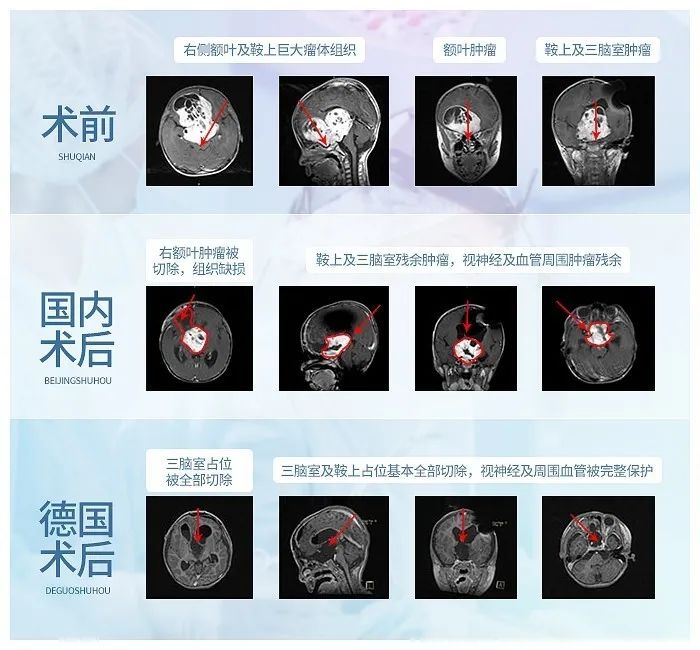
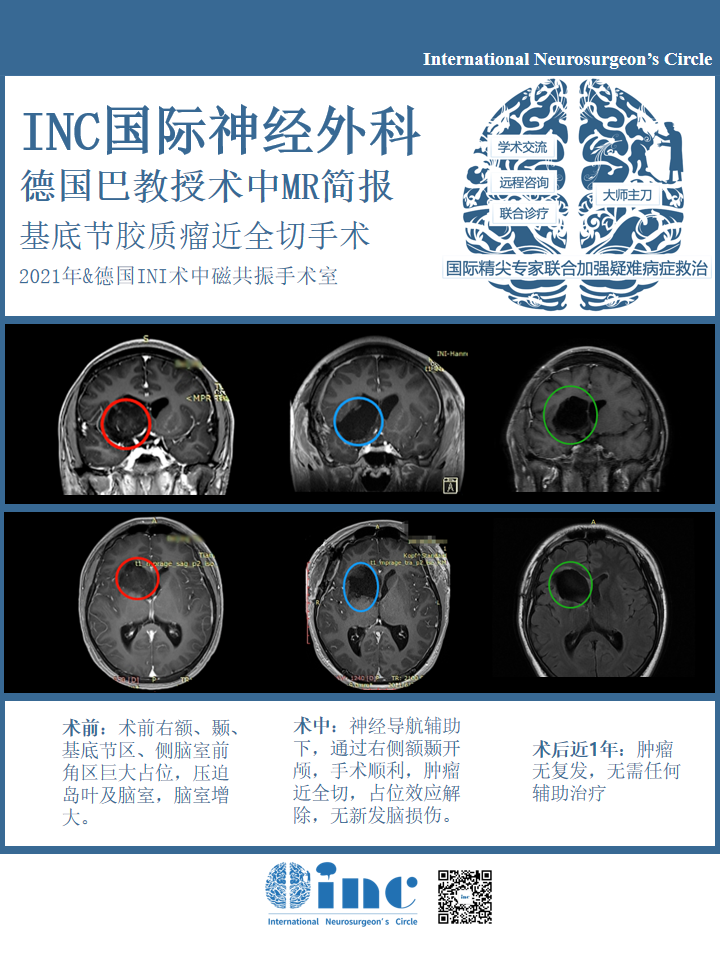
巨大基底节-岛叶胶质瘤赴德成功手术后1年
16岁少年圆梦远赴西藏
术前情况:16岁小磊(化名)在一次全身体检中查出“颅内占位性病变”,最后去国内知名三甲医院确诊为“右额、颞、基底节区、侧脑室前角区占位病变:胶质瘤可能性大……小磊的胶质瘤主要位于基底节部位,属于大脑功能区,目前手术风险极大,巨大占位压迫岛叶、和血管神经关系密切,切除肿瘤时极易造成脑组织神经血管损伤,90%以上会术后出现神经损伤如瘫痪、记忆障碍等,在这个位置手术不容有失,否则面临的可能是终身的瘫痪,生活不能自理。
治疗过程:为了给他最好的治疗,不辞辛苦竭尽全力找到世界盛名的大咖专家为他手术。同时也觉得自己太幸运了,得以遇到医术精湛的专家,在一场风险巨大极具手术挑战的手术中起死回生,术后的病理结果为预后良好的低级别胶质神经元性肿瘤,符合 WHO I 级胚胎发育不良性神经上皮瘤(DNET)。病变近全切除,未遗留神经损伤并发症如瘫痪、感觉障碍、记忆障碍等,肿瘤复发几率低,长期预后良好,可如常生活、学习。
手术一年的小磊不像一个经历过开颅手术的人。别人谈之色变的脑瘤,他已经恢复的和正常人没什么两样了,对于未来他充满动力。
术后第2天,小磊在康复师的指导下开始活动
术后1年的远程视频回访中,巴教授表示“我看了检查结果,非常开心,无复发,也不需要辅助放化疗,一切都很好!”
40岁刘先生脊髓髓内肿瘤,风险大易致残
术前情况:40岁刘先生因为出现持续的脖子肩膀酸痛、手抖、平衡性差、头晕等症状就医检查,诊断为脊髓髓内肿瘤,但是因为肿瘤长在解剖部位极其复杂的脊髓位置,手术瘫痪的风险极大,哪怕只是简单的活检,也可能会致残。多地资讯后都因为手术易致残建议不手术,但是不手术,症状只会越演越烈。

治疗过程:恰逢INC德国巴教授在国内进行示范手术,刘先生第一时间咨询了教授,结合刘先生的既往病史和影像资料,巴教授回复只要术中明确为肿瘤,那便是可以安全全切的,在国内医生的配合以及相关术中神经电生理监测等高科技设备的辅助下,巴教授主刀手术,于狭窄的方寸之内一层层剥离肿瘤,手术手法细致温柔,最终脊髓肿瘤被完整切除了下来。
术后情况:术后当天拔除气管插管,术后第2天即转出了ICU,术后第3天可下地自如行走。术后两周出院,患者术前症状大多已消失,无新发神经功能症状,无术后感染或并发症。目前无需其他辅助治疗。术后一年INC随访刘先生,他表示恢复良好,感谢巴教授给了他新生的机会。
巨大小脑胶质瘤严重压迫脑干
母子携手一场“生死相托”
术前情况:2020年末,6岁的辰辰在遭遇交通事故后,开始出现频繁头晕,就医检查发现小脑占位,提示低级别星形细胞瘤的可能,并且占位所在位置已压迫四脑室和脑干,中线结构局部左移,手术稍有不慎就可能出现瘫痪、长期昏迷等严重后果。最后,辰辰的父母选择向INC德国巴特朗菲教授寻求诊疗意见,教授表示:将尽力全切脑瘤。手术风险低于1-2%,如果确诊为毛细胞型星形细胞瘤,术后复发率将低于2-3%。
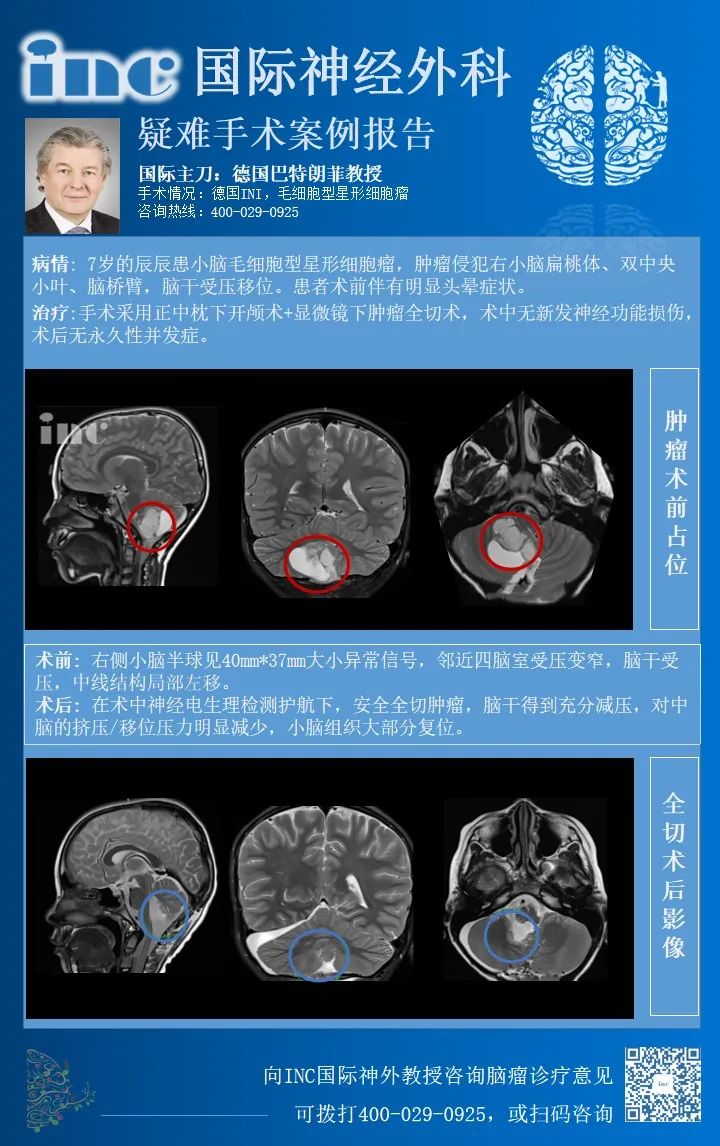
 患者术后病理报告
患者术后病理报告
2021年初,辰辰由母亲陪护赶赴德国接受教授手术治疗。在术中神经电生理检测护航下,巴教授安全全切肿瘤,术中无神经损伤,术后辰辰恢复状态很好。
回国后,辰辰的妈妈多次致谢INC,她说:"辰辰术后恢复状态很好,很多医生都看不出他做过手术,是INC和巴教授让孩子有机会继续感受这个世界的美好!“
9岁患儿丘脑胶质瘤全切获得治愈性疗效实录
患者情况:9岁的Dora正处于活泼好动的年纪,但她突然变得病恹恹的,不爱出门走动。父母本以为这是带她出门旅行过度疲劳造成的,简单休养即可恢复。可是几天后,Dora开始头痛、四肢无力、频繁呕吐,父母这才意识到问题的严重性,带她去医院检查,结果显示:右侧丘脑占位,医生建议尽快手术。 一想到孩子需要做开颅手术,Dora的父母就忧心不已,毕竟儿童与成年人的身体状况不同,手术必须由更为擅长小儿脑瘤治疗的专家做才稳妥。
治疗过程:怀着谨慎态度,他们来到北美知名儿童医院SickKids向James T. Rutka教授寻求第二诊疗意见。
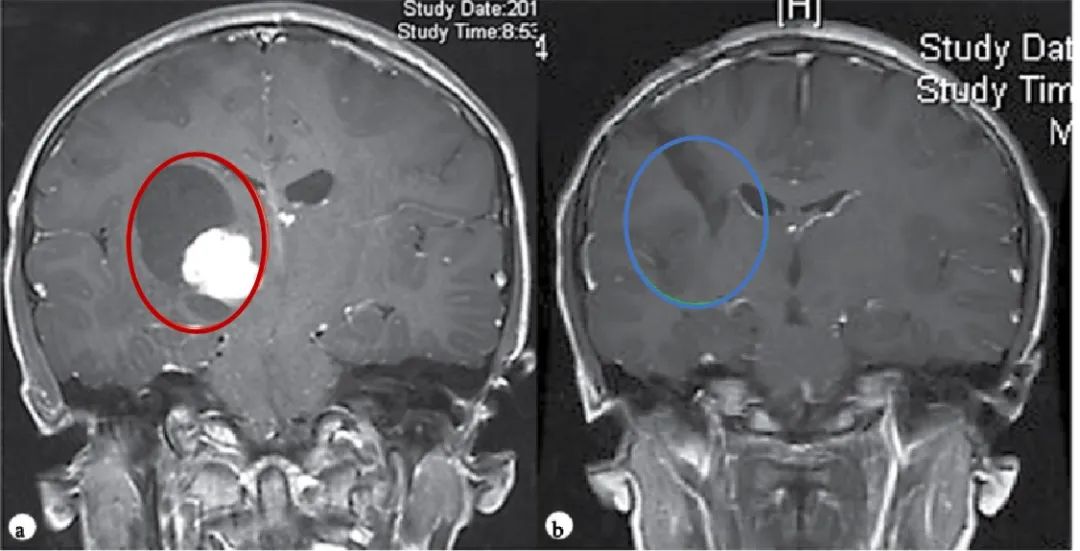
【图.术前(a)和术后(b)颅脑磁共振MR,术前MR显示右侧丘脑巨大囊实性占位,胶质瘤可能,皮质脊髓束(负责人体肢体运动的神经传导束)受压,脑室扩大,术后MR显示肿瘤全切,脑组织复位,无脑出血、脑水肿等损伤】
Rutka教授经诊断表示, Dora患上的是一种常见于儿童及青少年的良性胶质瘤——毛细胞星形细胞瘤。Rutka教授根据肿瘤生长的具体位置、形态特点,制定了安全的手术入路方案,并实现安全全切肿瘤,预后无并发症、无复发,对孩子的成长发育和智力没有造成任何影响。
INC是一个专注于世界神经外科领域技术超群、声誉斐然的大师级专家学术交流的医生集团。其成员教授为世界神经外科各专业的奠基者、开拓者,他们在各自领域对世界神经外科做出过巨大贡献,且其手术技术和能力在神经外科界有着难以替代的地位。INC一直致力国内外学术技术交流,也专注为国内追求更高质量手术、更佳预后的患者,提供世界前沿诊疗意见及世界级手术治疗。
自成立以来,在2018和2019的上海进博会期间,INC在上海外滩源(原英国总领事馆)开办两次世界规格的神经外科学术交流年会,INC的10多位来自不同国家不同地区的世界神经外科大师来华分享国外最新学术硕果、前沿技术和成功经验,堪称跨地域、无国界的国际神经外科学术盛宴。
: , 。 视频 小程序 赞 ,轻点两下取消赞 在看 ,轻点两下取消在看

