特应性皮炎是免疫性疾病还是细菌性疾病
时间:2022-11-30 15:00:26 热度:37.1℃ 作者:网络
一 什么是特应性皮炎?
特应性皮炎(atopic dermatitis,AD),又称特应性湿疹,是一种常见的炎症性皮肤疾病,其特征是反复出现湿疹样病变和强烈瘙痒,伴有显著的皮肤干燥[1]。过去30年全球范围内AD患病率逐渐增加,发达国家儿童AD患病率达10% ~ 20%,我国AD患病率的增加晚于西方发达国家和日本、韩国,但近10 年来增长迅速[1]。2016 年来自我国12 个城市的流行病学调查结果显示,1~7 岁儿童AD患病率已高达12.94%[2]。AD患者患哮喘、过敏性鼻炎、食物过敏和精神健康障碍的风险增加,且疾病的慢性复发过程给患者及其家属带来了极大的经济负担和较差的生活质量[2]。所以,了解AD独特的发病机制,并进行针对性的治疗,有助于改善患者症状,提高患者预后生存质量。
二 特应性皮炎的发病机制
AD的发病机制十分复杂,被认为与遗传和环境等因素关系密切[2]。父母亲等家族成员有过敏性疾病史是本病的强烈风险因素,遗传因素主要影响皮肤屏障功能与免疫平衡。本病患者往往有多种免疫学异常,其中Th2细胞的活化是其重要特征,还可有皮肤屏障功能破坏如表皮中丝聚蛋白减少或缺失。环境因素包括气候变化、生活方式改变、不正确的洗浴、感染原和变应原刺激等 [2]。虽然AD的确切发病机制尚不清楚,但目前研究认为,免疫异常、皮肤菌群紊乱、皮肤屏障功能障碍等因素是AD发病的重要环节[2] (如下图)。
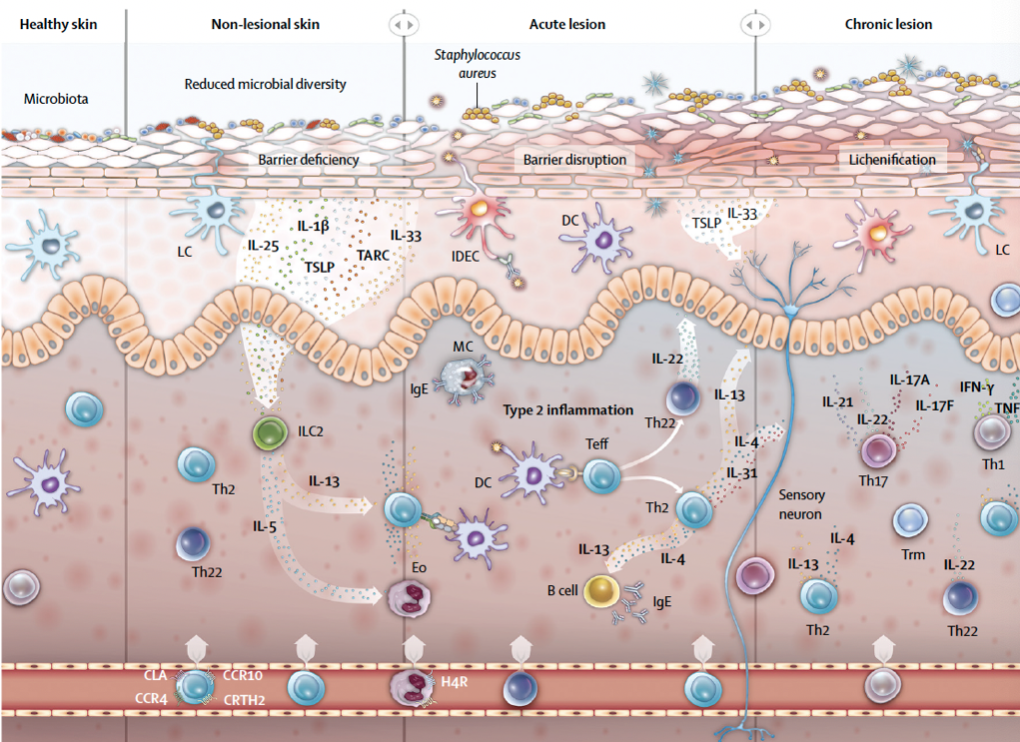
(引自Atopic dermatitis: Lancet 2020)
免疫调节紊乱和皮肤炎症是AD的发病机制的核心。AD患者的皮损显示出以CD4表达为主要特征的T细胞浸润。Th2型炎症是AD的基本特征,IL⁃4和IL⁃13是介导AD发病的重要细胞因子。AD炎症与表皮屏障功能破坏引起警报素释放,激活表皮树突状细胞和2型炎症反应相关。激活的Th2细胞释放IL-4和IL-13,促进B细胞的IgE类别转换,并通过信号转导和转录激活剂(STAT)途径产生抗原特异性IgE [1,3]。
AD的发生与菌群紊乱有关。研究发现,成人 AD 患者的皮损和非皮损区之间的细菌群落组成(β 多样性)显著不同,最大的差异是由于葡萄球菌种类的不同分布。三分之一的皮损区以金黄色葡萄球菌为主(相对丰度大于50%) [4]。一项出生队列研究发现,在出生后第2天存在金黄色葡萄球菌以外的其他葡萄球菌的定植,可能会保护婴儿免受AD的影响[5]。
在AD患者的皮损和非皮损区均可见表皮屏障功能障碍,表现为经表皮失水增加、pH值升高和脂质成分改变。表皮屏障会因微生物失调而进一步被破坏,其中包括金黄色葡萄球菌和马拉色菌的定植。受到刺激的应激表皮屏障中,其角化细胞会通过表皮警报素IL-33和胸腺基质淋巴生成素(TSLP)释放促炎和致瘙痒信号,驱动2型炎症细胞的募集,激活皮肤中的2型固有淋巴样细胞。而这些淋巴样细胞会产生IL-5和IL-13,激活嗜酸性粒细胞和Th2细胞,导致炎症发生[1,3]。
如上所述,AD的发病机制十分复杂,包括表皮屏障障碍、菌群紊乱和主要的2型免疫调节失调。而相较于单一因素,这些驱动因素更多的是与其他因素相互作用促进AD的发生。例如,IL-31是由T细胞产生,能引起强烈瘙痒的细胞因子,IL-31及其受体在皮损处都过度表达,葡萄球菌的肠毒素能够上调IL-31的表达;搔抓能够增强金黄色葡萄球菌和皮肤的结合;金黄色葡萄球菌产生的神经酰胺酶会加剧皮肤屏障的损伤,诱发炎症反应;丝聚蛋白缺乏导致的皮肤屏障障碍促进炎症和T细胞浸润;局部Th2免疫反应会进一步削弱屏障功能,刺激瘙痒,促进菌群失调,有利于葡萄球菌属成员,特别是金黄色葡萄球菌定植。
三 金黄色葡萄球菌在特应性皮炎中的作用
金黄色葡萄球菌是 AD 发展过程中微生物研究的主要菌种。这种革兰氏阳性细菌寄生于呼吸道和皮肤。金黄色葡萄球菌通常在 AD 患者的皮肤上发现,根据患者的年龄,AD 的严重程度,样本量以及检测金黄色葡萄球菌的采样和分析方法,报告的检出率从30% 到100% 不等,而在健康对照中,检出率约为20%。研究发现金黄色葡萄球菌的定植数目和病原体密度与AD疾病的严重程度有关。在 AD 患者严重发作期间,微生物群内葡萄球菌属的组成可降低为单一菌株的金黄色葡萄球菌。而在治疗和恢复期间,微生物组成恢复到发作前的组成[1,3,5]。金黄色葡萄球菌定植可能是复发的直接原因。[13]
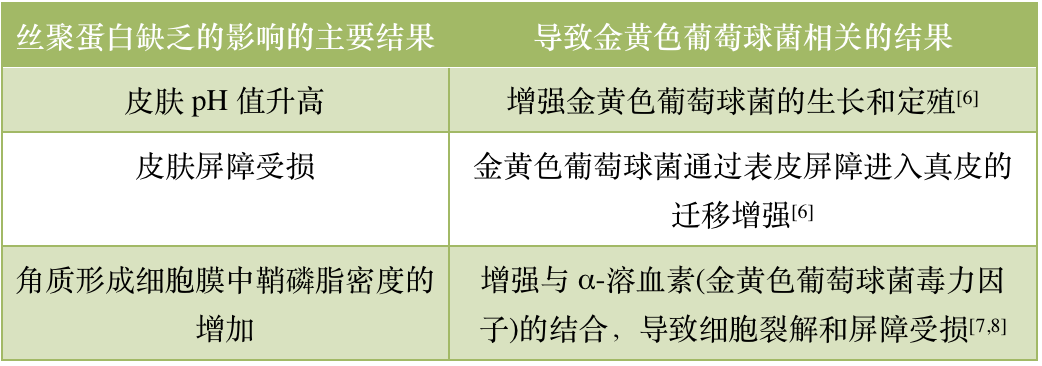
AD 患者丝聚蛋白的缺陷既增加了金黄色葡萄球菌的定植,又增强了金黄色葡萄球菌介导的细胞毒性和免疫激活[6-8](表1)。丝聚蛋白的分解产物尿酚酸(UCA)和吡咯烷酮羧酸(PCA)会导致皮肤酸化,而AD 患者由于丝聚蛋白的缺乏,其产物UCA和 PCA也同时缺乏,从而引起皮肤 pH 变化。而更中性的pH值和定植的金黄色葡萄球菌增加有关,导致AD 患者皮肤金黄色葡萄球菌的生长和定植增加[6]。此外,金黄色葡萄球菌会分泌一种毒力因子——α-溶血素。α-溶血素能粘附在角质形成细胞膜的鞘磷脂上,导致细胞裂解和皮肤屏障破坏。丝聚蛋白缺乏以及 Th2细胞因子增加会下调酸性鞘磷脂酶,从而增强α-溶血素和鞘磷脂的结合效率,加重细胞裂解和屏障受损[7,8]。
表 1|丝聚蛋白缺乏对金黄色葡萄球菌皮肤定植及毒力的影响
有研究支持这样的假设:金黄色葡萄球菌的毒力可能是 AD 恶化的主要驱动因素,甚至可能是AD 加重的直接原因[7-12](表2)。研究发现,相较于健康人群的皮肤,AD患者皮肤分离的金黄色葡萄球菌毒力因子上调,如PSM-a[9]、δ毒素[10]、α-溶血素[7,8]、肠毒素 B[12]和聚集因子 B[12]等。一项小鼠实验发现,金黄色葡萄球菌会通过PSM-a的分泌,在小鼠体内促进白细胞介素(IL)-17A的释放,促进炎症反应,导致小鼠皮肤炎症和屏障破坏[9]。而另一项关于δ毒素的小鼠研究也发现,金黄色葡萄球菌产生的δ毒素可能通过诱导肥大细胞脱颗粒、IgE 释放和 IL-4表达增加介导炎症反应[10]。相较于健康皮肤,研究发现来自AD皮肤的金黄色葡萄球菌更容易携带编码肠毒素的基因,并且更容易产生这些毒素[11]。肠毒素B被证明可能是通过增强的 T 细胞信号介导炎症,最终在AD 患者中引起红斑和表皮增厚[11]。同样相较于健康皮肤,从 AD 皮肤分离的金黄色葡萄球菌具有增强的凝集因子 B 的结合活性,从而增加了其对角质细胞的粘附,使得金黄色葡萄球菌的定植增加[12]。
表 2|参与 AD的金黄色葡萄球菌毒力因子

综上所述,AD患者中的皮肤屏障功能受损,pH 值升高和皮肤炎症等,可以促进皮肤微生物群落的变化。而金黄色葡萄球菌可以诱导皮肤炎症和加重 AD。因此,在AD中,可能存一个恶性循环:AD皮肤中的丝聚蛋白缺乏,会导致金黄色葡萄球菌的定植增强,然后其通过表达毒力因子,诱导皮肤炎症并导致进一步的皮肤屏障损伤,进一步促进维持不平衡的皮肤微生物群落(如下图)。
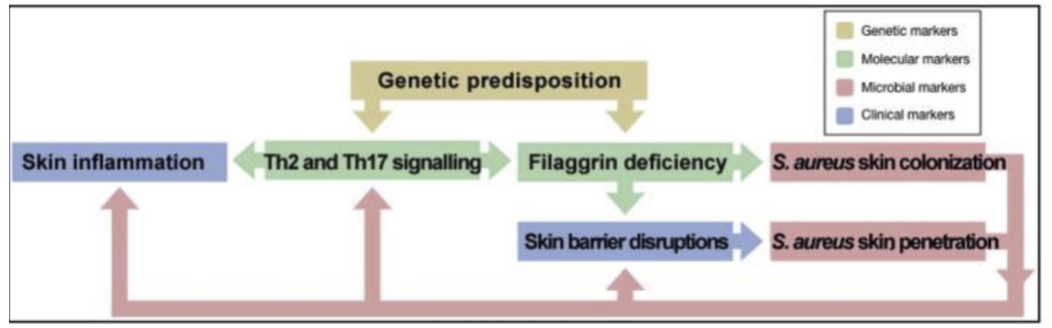
(引自Skin Microbiome in Atopic Dermatitis: Acta Derm Venereol.2020)
四 总结
AD全年龄均可发病,多伴有剧烈的反复性瘙痒表现。AD疾病的慢性复发过程和合并症的发生,给患者及其家属带来了极大的经济负担和较差的生活质量。结合其疾病特点、症状表现和发病机制,临床需积极开展免疫治疗和抗菌治疗,从而改善患者的相关临床症状,提高临床疗效,改善特应性皮炎患者的预后及生活质量。
参考文献
1.Langan SM, Irvine AD, Weidinger S. Atopic dermatitis [published correction appears in Lancet. 2020 Sep 12;396(10253):758]. Lancet. 2020;396(10247):345-360. doi:10.1016/S0140-6736(20)31286-1
2.中华医学会皮肤性病学分会免疫学组, 特应性皮炎协作研究中心. 中国特应性皮炎诊疗指南(2020版)[J]. 中华皮肤科杂志, 2020, 053(002):81-88.
3.Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. 2018;4(1):1. Published 2018 Jun 21. doi:10.1038/s41572-018-0001-z
4.Edslev SM, Agner T, Andersen PS. Skin Microbiome in Atopic Dermatitis. Acta Derm Venereol. 2020 Jun 9;100(12):adv00164. doi: 10.2340/00015555-3514. PMID: 32419029; PMCID: PMC9189751.
5.Sroka-Tomaszewska J, Trzeciak M. Molecular Mechanisms of Atopic Dermatitis Pathogenesis. Int J Mol Sci. 2021 Apr 16;22(8):4130. doi: 10.3390/ijms22084130. PMID: 33923629; PMCID: PMC8074061.
6.Miajlovic, Helen et al. “Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus.” The Journal of allergy and clinical immunology vol. 126,6 (2010): 1184-90.e3. doi:10.1016/j.jaci.2010.09.015
7.Brauweiler, Anne M et al. “Th2 cytokines increase Staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (STAT6).” The Journal of investigative dermatology vol. 134,8 (2014): 2114-2121. doi:10.1038/jid.2014.43
8.Brauweiler, Anne M et al. “Filaggrin-dependent secretion of sphingomyelinase protects against staphylococcal α-toxin-induced keratinocyte death.” The Journal of allergy and clinical immunology vol. 131,2 (2013): 421-7.e1-2. doi:10.1016/j.jaci.2012.10.030
9.Williams, Michael R et al. “Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis.” Science translational medicine vol. 11,490 (2019): eaat8329. doi:10.1126/scitranslmed.aat8329
10.Nakamura, Yuumi et al. “Staphylococcus δ-toxin induces allergic skin disease by activating mast cells.” Nature vol. 503,7476 (2013): 397-401. doi:10.1038/nature12655
11.Skov, L et al. “Application of Staphylococcal enterotoxin B on normal and atopic skin induces up-regulation of T cells by a superantigen-mediated mechanism.” The Journal of allergy and clinical immunology vol. 105,4 (2000): 820-6. doi:10.1067/mai.2000.105524
12.Fleury, Orla M et al. “Clumping Factor B Promotes Adherence of Staphylococcus aureus to Corneocytes in Atopic Dermatitis.” Infection and immunity vol. 85,6 e00994-16. 23 May. 2017, doi:10.1128/IAI.00994-16
13.[Chng KR, et al. Nat Microbiol. 2016; 1: 16106]
