Cardiovasc Res 天津医科大学朱毅教授团队揭示内皮METTL3-RUNX1-eNOS信号通路在血压调控中的作用机制
时间:2024-11-15 13:00:09 热度:37.1℃ 作者:网络
高血压是导致心血管发病率和死亡率最主要的风险因素之一,其显著特征是血管功能受损,并与内皮功能障碍密切相关。内皮细胞(Endothelial cells, ECs)作为循环血液与血管壁内皮下组织的屏障,可以产生多种调节血管活性物质包括舒张和收缩因子来维持血管生理稳态。N6-甲基腺苷(N6-methyladenosine, m6A)是真核生物mRNA中最丰富的转录后修饰,这种可逆的RNA甲基化修饰受到多种酶的调节,包括甲基转移酶(Writers)、去甲基化酶(Erasers)和m6A甲基化识别蛋白(Readers),其中甲基转移酶样3 (Methyltransferase-like 3, METTL3)作为具有催化活性的核心亚基发挥作用。然而,内皮细胞METTL3在血压调控中的作用及其潜在机制仍不清楚。
2024年11月12日,天津医科大学基础医学院朱毅、李博川团队与杨扬团队在Cardiovascular Research杂志上在线发表了题为“Regulation of Blood Pressure by METTL3 via RUNX1b-eNOS Pathway in Endothelial Cells”的研究论文,揭示了METTL3介导的m6A甲基化修饰在内皮细胞介导的血压调控机制中的作用,为高血压的防治提供了新的干预靶点。

首先,研究者使用Ang Ⅱ建立了高血压小鼠模型。体内和体外的结果显示,METTL3在内膜中的表达高于在中膜中的表达,并在Ang Ⅱ处理后显著降低,而在其他m6A调节因子中没有观察到显著改变。同时,Ang Ⅱ处理后,内膜p-eNOS和t-eNOS以及血清NO水平下降,而内皮细胞中表现出METTL3和eNOS蛋白水平的降低,但不影响其他m6A调节因子。这些结果提示,在Ang Ⅱ诱导的高血压模型中,METTL3/eNOS信号通路表达降低。
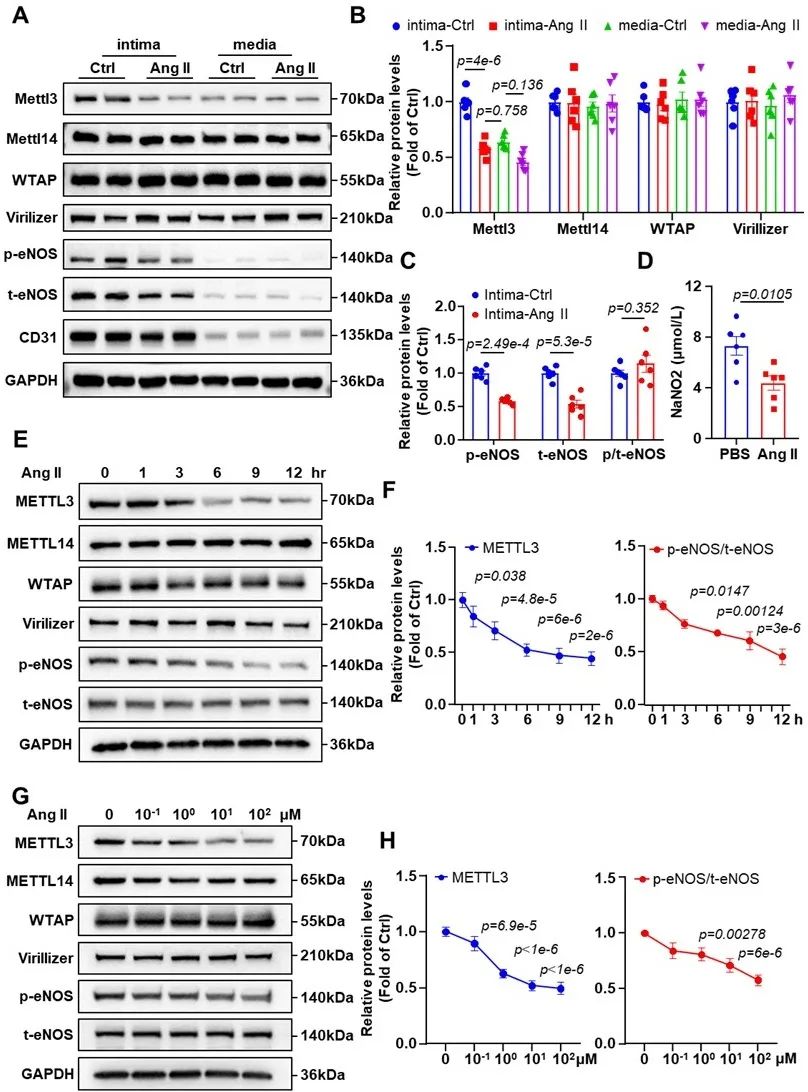
图1. METTL3在Ang Ⅱ诱导的高血压小鼠模型中降低
进一步,研究者通过使用内皮特异性METTL3敲除(EC-Mettl3KO)小鼠与对照小鼠相比,EC-Mettl3KO小鼠在基础水平和受到Ang Ⅱ刺激的反应中表现出SBP和DBP增加。此外,通过对EC-Mettl3KO小鼠的血管环实验、免疫印迹和一氧化氮(NO)含量检测实验发现,NO介导的血管舒张反应在内皮特异性敲除METTL3导致的内皮功能障碍与血压调节中起着关键作用。
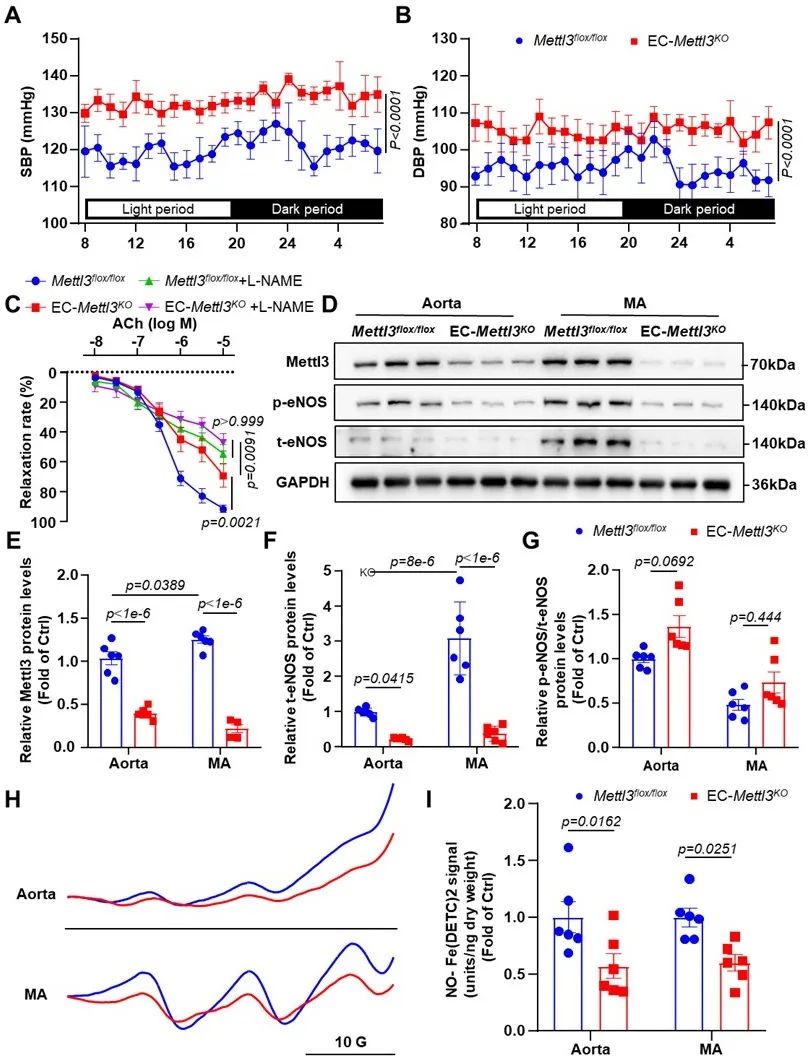
图2. 内皮特异性METTL3敲除小鼠通过抑制eNOS升高血压
机制上,通过对MeRIP-seq分析发现,编码eNOS的基因NOS3的甲基化状态并没有受到m6A甲基化修饰的直接调控。RUNX1 mRNA水平对Ang Ⅱ和METTL3敲低反应显著增加,而METTL3的过表达显著逆转了Ang Ⅱ诱导的RUNX1 mRNA水平的增加。因此,体内和体外的结果共同证实了Ang Ⅱ诱导的内皮细胞激活是由RUNX1上的m6A修饰所介导的。为了验证eNOS是否是RUNX1的直接靶基因,研究者通过染色质免疫沉淀(ChIP)实验结果证实了RUNX1和eNOS启动子之间的直接联系。使用选择性RUNX1-CBFβ相互作用抑制剂Ro5-3335改善了EC-Mettl3KO小鼠的内皮依赖性血管舒张功能以及si-METTL3内皮细胞中eNOS和NO的表达水平。这些结果表明eNOS是RUNX1的新的直接下游靶基因,并且RUNX1的抑制改善了由METTL3缺失引起的eNOS表达的降低。
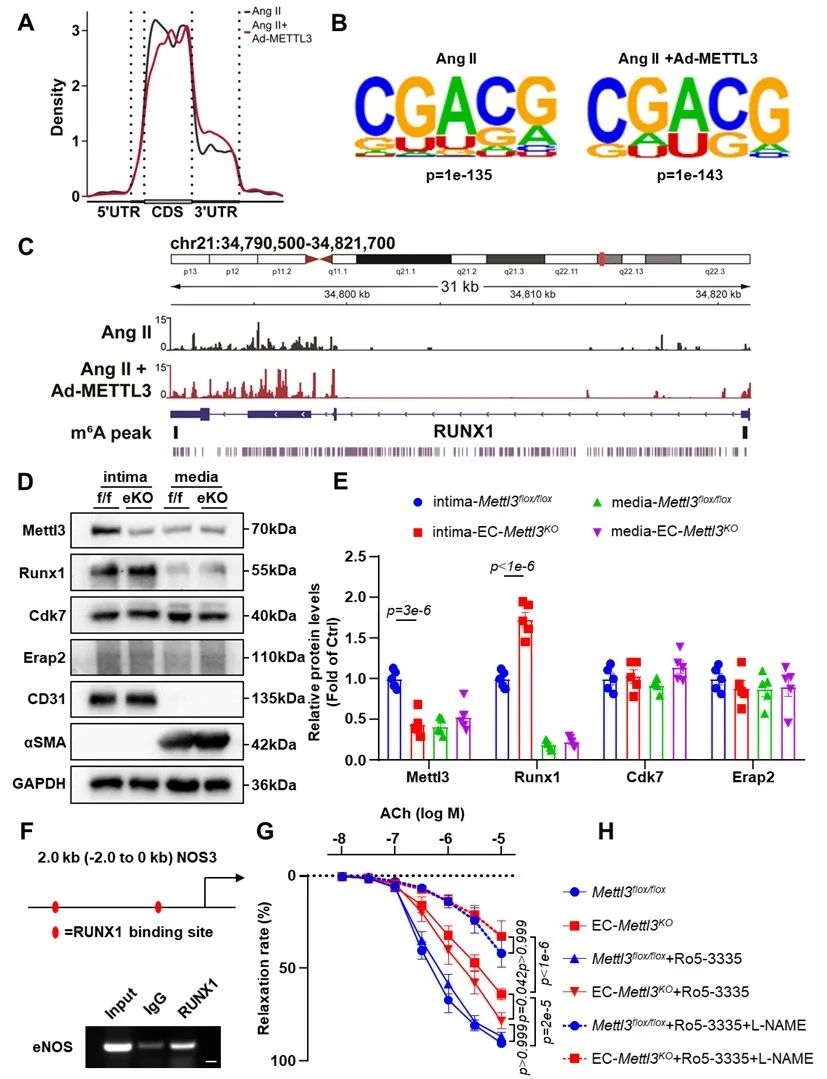
图3. METTL3介导RUNX1的m6A甲基化
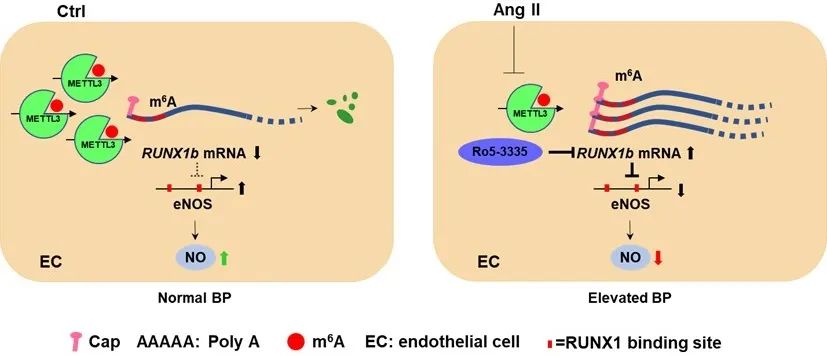
综上,在正常的内皮细胞中,METTL3催化m6A甲基化修饰的形成,从而促进RUNX1b mRNA降解,维持eNOS表达及NO含量。而当细胞感受到Ang Ⅱ刺激时,METTL3减少导致RUNX1b的降解效率降低,eNOS表达受抑制,NO产生水平降低,导致内皮细胞激活,进而升高血压。
天津医科大学基础医学院博士研究生张彦宏和杨晓晓为共同第一作者。李博川副教授、朱毅教授和杨扬教授为文章的共同通讯作者。上述工作得到了国家自然科学基金重点项目和面上项目的资助。
原文链接:
https://academic.oup.com/cardiovascres/advance-article-abstract/doi/10.1093/cvr/cvae242/7895714?redirectedFrom=fulltext&login=false

