病例报告|PRRT2基因c.776del突变致发作性运动诱发性运动障碍1例
时间:2024-08-23 22:01:45 热度:37.1℃ 作者:网络
摘 要 报道1例典型的发作性运动诱发性运动障碍(paroxysmal kinesigenic dyskinesia, PKD)患者。该患者为26岁女性,病史10年,表现为安静状态下突然运动出现发作性四肢和头部舞蹈样动作,无感觉先兆,持续数十秒后自行缓解,发作时和发作间期意识清醒,存在明显家族史,神经系统查体正常,头颅磁共振及脑电图未见异常,基因检测结果显示,先证者及其有相似表型的父亲存在PRRT2基因c.776del移码突变。根据PKD诊断标准,该患者可确诊为PKD,经奥卡西平治疗1个月后症状明显缓解,预后较好。PKD为罕见的运动障碍疾病,该患者症状典型,其基因突变位点在人类基因突变数据库中尚未报道,因此,本文丰富了PKD致病基因突变谱,为PKD遗传咨询提供依据,同时可增加临床医生对该病的认知。
关键词
发作性运动诱发性运动障碍;运动障碍性疾病;移码突变;临床表现;PRRT2
发作性运动诱发性运动障碍(paroxysmal kinesigenic dyskinesia, PKD)是一种罕见的以运动诱发短暂的不自主运动为特征的神经系统疾病,又称为发作性运动诱发性舞蹈手足徐动症,临床上表现为静止状态下因突然运动而诱发的不自主运动,包括肌张力障碍、舞蹈症、手足徐动症或多种症状组合发生,常被误诊为癫痫或其他发作型疾病[1]。绝大多数PKD为原发性,PRRT2是PKD的主要致病基因,目前已经报道约80种致病变异,不同变异的临床表现具有高度异质性。因此,不断筛选和总结新的PKD变异及其临床表现具有重要意义[2]。现报道中山大学附属第一医院门诊诊治的1例PRRT2基因突变的PKD患者,其致病位点为首次报道。
1 临床资料
患者,女,26岁,主因“发作性四肢不自主抖动10余年”于2023年7月就诊于我院神经科门诊。患者于2013年开始出现发作性四肢和头部不自主运动,呈手舞足蹈样动作,发作前无肢体发麻、发凉、发冷等不适。安静状态下突然运动容易出现,起身、过马路以及紧张情境下易出现,表现为头向后仰随即倒地,发作时和发作间期意识清醒,每天均有发作,每次持续数十秒后自行缓解。患者无明显肢体麻木与疼痛,日常生活可自理,未影响正常生活,当地医院尚未诊治。自发病以来食欲尚可,睡眠一般,压力不大,大小便正常,体质量无明显变化。既往史与个人史:自幼患荨麻诊,无其他疾病病史,既往顺产,无脑外伤病史,无婴儿惊厥病史,按计划接种疫苗,否认特殊药物和毒物接触史。家族史:父亲、弟弟有类似发作性症状(图1)。
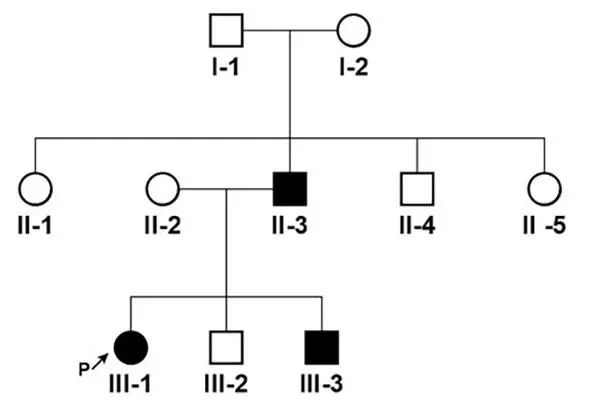
图1 患者家系图 Ⅲ-1为先证者Fig.1 Family map of the patient
体格检查:神清,全身可见散在荨麻疹,其余内科查体未见明显异常,专科查体未见明显异常。辅助检查:患者发作间期脑电图未见明显异常,颅脑磁共振及血管成像显示右侧颈内动脉C7段小动脉瘤,其余未见明显异常。实验室检查:促甲状腺刺激激素(TSH)0.012 μIU/mL(参考值:0.560~5.910 μIU/mL ),游离甲状腺素T3 13.6 pmol/mL(参考值:3.810~6.910 pmol/mL ),游离甲状腺素T4 41.6 pmol/mL(参考值:7.500~21.100 pmol/mL )。全外显子测序显示该患者PRRT2基因发生一处变异:NM_145239.3:c.776del(p.Gly259Valfs*54),根据ACMG指南,该变异初步判定为可能致病性变异,经查阅文献及数据库,该变异未曾报道。经Sanger测序家系验证,提示变异来自父亲(图2)。
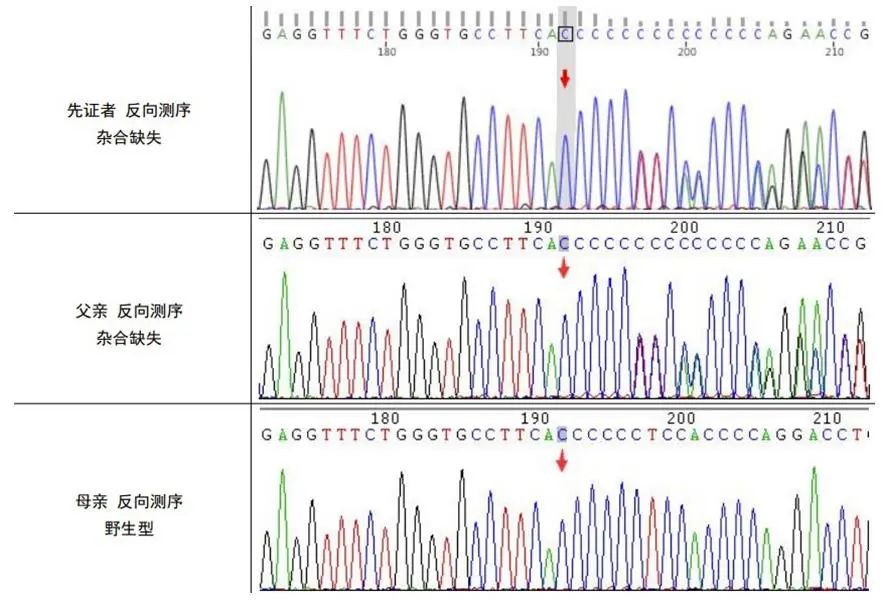
图2 患者及其父母Sanger测序结果 该基因突变来源于父亲Fig.2 Sanger sequencing results of the patient and her parents
本例患者表现为发作性四肢不自主运动,主要由运动及紧张情绪诱发,发作时间较短,可自行缓解,发作期和发作间期无意识障碍,神经系统查体正常,结合明显家族史及PRRT2基因突变,可确诊发作性运动诱发性运动障碍。给予奥卡西平(0.3 g,每日2次)治疗。2023年9月及2024年2月随访患者发作次数明显减少,症状明显减轻。
2 讨论
本例患者呈发作性起病,慢性进展,主要临床特点:①发作性不自主运动,由安静时突然运动或起身、紧张等诱发;②不自主运动主要表现为头部及四肢扭曲样形态;③发作时间较短,持续约数十秒,可自行缓解,发作期及发作间期无意识障碍;④发病年龄为16岁;⑤神经系统查体未见明显异常,脑电图及颅脑磁共振未发现明显异常,可排除器质性病变及癫痫发作;⑥具有明显家族史;⑦基因检测存在PRRT2基因杂合可疑致病突变,根据BRUNO等提出的PKD诊断标准,可诊断为发作性运动诱发性运动障碍。
发作性运动障碍是一组以不同病因引起的不自主运动间歇性发作为特征的神经系统疾病,PKD是最常见的发作性运动障碍,男性发病率是女性的2~4倍,年轻患者较为多见,有研究指出大约87%的患者发病年龄7~15岁[3]。按照病因,PKD可以分为原发性和继发性2种类型,绝大多数具有家族遗传倾向,主要以常染色体显性方式遗传,PKD可伴有不完全外显,并可能存在遗传早现现象[4]。本例为女性,发病年龄稍晚,临床上较为少见,此外也有老年起病PKD患者的报道[5],提示PKD在发病年龄上存在异质性。PKD突出性特征是运动诱发,发作时多表现为肌张力障碍、舞蹈样动作以及投掷样动作。对于家族性PKD,通常累及双侧肢体,表现为复杂的四肢舞蹈样动作,而散发性PKD通常表现为病程较短的单侧肌张力障碍。先兆是PKD另一个突出的临床表现,指突然运动引起不自主运动前的异常感觉,主要表现为受累肢体的麻木、疼痛或肌无力等[6],而对于某些症状较轻的患者,感觉先兆之后并未出现运动障碍发作[7]。本例常因安静时突然运动而诱发,多表现为肌张力障碍和舞蹈样动作,在发病前并未相关感觉异常,临床表现较为典型,符合家族性PKD的特点。
PKD发病机制尚不明确,运动皮质或基底节功能障碍是可能的致病机制,并与基因突变相关,最常见的致病基因为PRRT2和TMEM151A[8]。PRRT2基因于2011年首次证实,定位于16p11.2,包含4个外显子,编码340个氨基酸,目前已报道致病位点约90个(包括不完全外显),主要为移码变异,无义变异以及错义变异。PRRT2导致PKD的具体机制目前尚未阐明,可能和神经系统发育、蛋白错误定位以及离子通道功能障碍有关[9]。研究表明PRRT2蛋白在大脑皮层、基底神经节以及小脑中高表达,通过神经元突触前蛋白调节突出囊泡的融合,进而调节神经递质释放,PRRT2基因突变会导致突触传递功能障碍,并可能引起小脑平行纤维-小脑浦肯野细胞突出传递异常,从而导致发病[10]。
本例突变位点为NM_145239.3:c.776del(p.Gly259Valfs*54),是PRRT2基因编码区因非三倍数碱基缺失导致的移码突变,为最常见的变异类型,经查阅文献及大规模人群频率数据库gnomAD,未发现该变异报道,因此该位点为PRRT2基因首次发现的新致病位点。PRRT2基因移码突变可通过mRNA降解或编码氨基酸序列的提前终止,影响mRNA稳定性,从而引起正常蛋白功能的丧失[11]。PKD移码突变较为常见,研究发现大约78.5%的患者携带相同的移码突变(c.649dupC;p.Arg217Profs*8),该位点所在的序列具有特殊的连续9个胞嘧啶碱基的特殊结果,其可能容易形成发卡结构并导致DNA聚合酶滑脱,从而引起相关移码突变[12]。
由于PKD发病率较低且临床表现复杂,易误诊。PKD发作形式具有重复性、短暂性及刻板性的特征,对抗癫痫药物敏感,且部分患者婴幼儿时期曾出现惊觉发作,因此常被误诊为癫痫发作,临床上需加以鉴别。PKD的治疗包括药物治疗和非药物治疗,是否药物治疗以及药物的选择和剂量需要结合患者发病年龄、临床表现以及治疗意愿综合考虑,药物治疗推荐首选卡马西平或奥卡西平,其次为拉莫三嗪或苯妥英钠[1]。研究发现对于PRRT2基因突变患者,卡马西平治疗反应效果较强[13]。本例使用奥卡西平治疗,经半年随访,其症状改善明显,预后较好,与文献报道一致。
参考文献:
1. CAO L, HUANG X, WANG N, et al. Recommendations for the diagnosis and treatment of paroxysmal kinesigenic dyskinesia: an expert consensus in China[J]. Transl Neurodegener, 2021, 10(1): 7.
2. HUANG X J, WANG S G, GUO X N, et al. The Phenotypic and Genetic Spectrum of Paroxysmal Kinesigenic Dyskinesia in China[J]. Mov Disord, 2020, 35(8): 1428-1437.
3. 刘晓黎, 詹飞霞, 黄啸君,等. 携带PRRT2基因c.649dupC突变的发作性运动诱发性运动障碍临床表型总结[J]. 中国神经精神疾病杂志, 2022, 48(4): 206-212.
4. 毕光辉, 曲星华, 张慧芳, 等. 发作性运动诱发性运动障碍一家系临床与遗传学特点 [J]. 中国神经精神疾病杂志, 2016, 42(4): 216-221.
5. YAO L, LIANG W, MEI S, et al. Elderly-Onset Paroxysmal Kinesigenic Dyskinesia: A Case Report[J]. Neurol Ther, 2022, 11(4): 1805-1811.
6. BHATIA K P. Paroxysmal dyskinesias [J]. Mov Disord, 2011, 26(6): 1157-1165.
7. MENERET A, GRABLI D, DEPIENNE C, et al. PRRT2 mutations: a major cause of paroxysmal kinesigenic dyskinesia in the European population[J]. Neurology, 2012, 79(2): 170-174.
8. XU J J, LI H F, WU Z Y. Paroxysmal Kinesigenic Dyskinesia: Genetics and Pathophysiological Mechanisms[J]. Neurosci Bull, 2023. doi: 10. 1007/S12264-023-01157-2.
9. 刘鹏鹏, 刘亚青, 令调文, 等. PRRT2基因突变致病机制与相关疾病的研究进展[J]. 中国神经精神疾病杂志, 2020, 46(6): 361-364.
10. TAN G H, LIU Y Y, WANG L, et al. PRRT2 deficiency induces paroxysmal kinesigenic dyskinesia by regulating synaptic transmission in cerebellum [J]. Cell Res, 2018, 28(1): 90-110.
11. PAN Y, LIU Q, ZHANG J, et al. PRRT2 frameshift mutation reduces its mRNA stability resulting loss of function in paroxysmal kinesigenic dyskinesia [J]. Biochem Biophys Res Commun, 2020, 522(3): 553-559.
12. EBRAHIMI-FAKHARI D, SAFFARI A, WESTENBERGER A, et al. The evolving spectrum of PRRT2-associated paroxysmal diseases [J]. Brain, 2015, 138(Pt 12): 3476-3495.
13. LI H F, CHEN W J, NI W, et al. PRRT2 mutation correlated with phenotype of paroxysmal kinesigenic dyskinesia and drug response [J]. Neurology, 2013, 80(16): 1534-1535.
【引用格式】黄森,钟展华,陈伟能,等. PRRT2基因c.776del突变致发作性运动诱发性运动障碍1例[J]. 中国神经精神疾病杂志,2024,50(4):239-241.
【Cite this article】HUANG S,ZHONG Z H,CHEN W N,et al. Paroxysmal motor induced dyskinesia caused by PRRT2 gene c.776del mutation[J]. Chin J Nervous Mental Dis,2024,50(4):239-241.
DOI:10.3969/j.issn.1002-0152.2024.04.008

