Nat Commun:李昊等开发人工智能计算方法CGMega解析癌症基因模块
时间:2024-07-21 12:00:44 热度:37.1℃ 作者:网络
多基因协同互作——细胞微观世界内的基本策略
单个受精卵如何构建起复杂的生命“楼宇”?肿瘤如何成为生命的最大“宿敌”?生命又为何会走向衰老?细胞,作为生命的最基本单位,无疑是解答这些问题的最佳起点。历经一个多世纪的探索,科学家们已成功见证了这个曾经看似不可企及的世界。微观下的人类细胞世界仿佛一座偌大的城市,处处呈现着“秩序”与“协调”,2米长的DNA序列有序地缠绕在微米级别的细胞核内,架起一座座“立交桥”形成染色质环、拓扑关联结构域等复杂空间结构,保障着细胞内基因信息的精准、高效传播。
然而,基因组结构如何维持动态有序、生物信息如何协同互作等一直以来是科学家们迫切渴望解读的生命“奥秘”,传统手段如还原论等仅聚焦单个基因或分子,然而,一个活细胞的复杂功能是通过许多基因及其产物的协同活动来完成的[1],多基因协同互作才是细胞活动的基本策略。从复杂控制系统的视角来看,由多基因协同互作所形成的基因模块是细胞活动的基本控制单元。表观遗传修饰扮演控制信号,在基因组三维结构的信息传播路径下,调控基因的表达,最终影响细胞的生理和病理过程。
人工智能技术——破译多组学视角下癌症基因模块的利器
大量研究表明,疾病相关基因并不是随机分布在生物网络中,相反,他们往往聚集形成疾病基因模块,这种现象在癌症中更为突显(图1)[2-4]。解读癌症基因模块对于肿瘤形成机制解析[5-6]、肿瘤精准分型[7]、抗肿瘤药物筛选[8]等方向具有重要意义,采用计算方法从海量组学数据中建模预测已成为基因模块研究的主要手段之一[9,10]。然而,高维度、跨尺度生物数据的高效整合以及高阶拓扑关系的辨识仍是基因模块计算分析方法面临的严峻挑战。
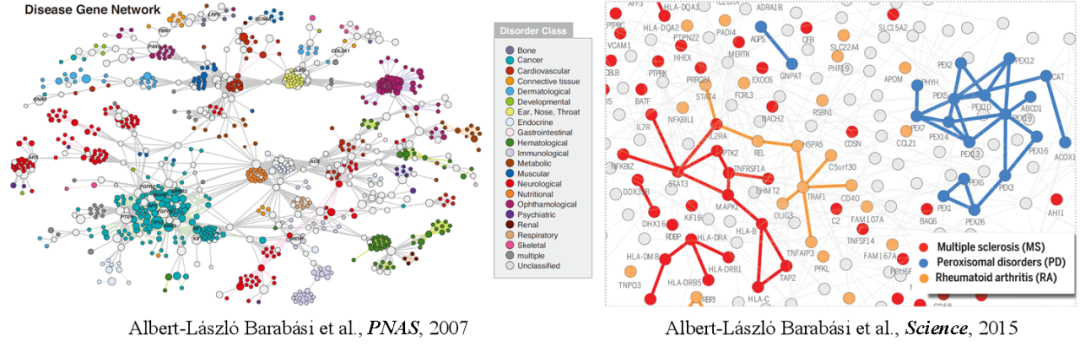
图1. 基因模块在疾病中广泛存在
近期,针对癌症基因模块识别问题中面临的多模态信息融合、小样本学习、子图搜索等计算挑战,军事医学研究院伯晓晨、陈河兵团队联合上海交通大学杨旸团队提出基于可解释图注意力机制的计算框架CGMega(图2),实现了癌症基因模块的辨识与解析。CGMega是一个基于Transformer的图注意力网络模型,其任务性质为半监督的图节点分类学习。输入是由蛋白质互作、基因组、表观基因组以及三维基因组等多组学信息融合的图,该图以基因为节点,以蛋白质互作关系为相应节点的边,将预处理后的染色质结构信息、变异信息、拷贝数信息、染色质开放信息以及组蛋白修饰信息分配到各个基因作为节点特征。CGMega的输出是对于每个基因的癌基因预测概率。最后,CGMega采用并改进GNNExplainer可解释工具来进行癌症基因模块的挖掘,通过掩蔽的方法确认子图结构以及节点特征对于关键癌基因的贡献程度。
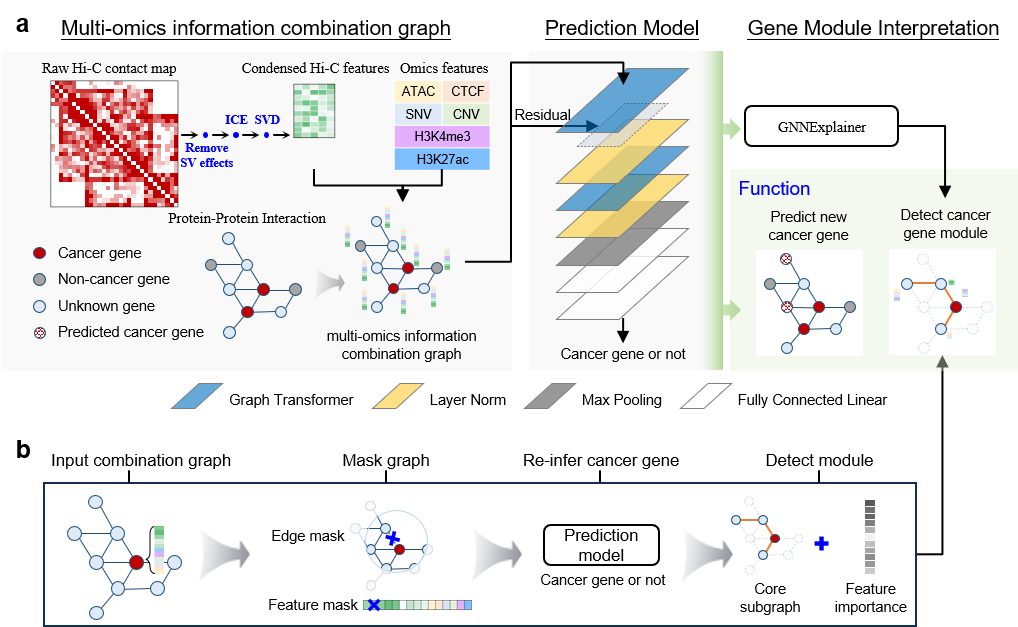
图2. CGMega计算框架图
团队在乳腺癌MCF7细胞系对CGMega进行了性能测试,预测结果为AUPRC=0.9140,AUROC=0.9630。团队对CGMega与其他方法进行了比较,包括4种通用模型GCN, GAT, MLP, SVM以及3种针对癌基因预测的领域模型如MTGCN、EMOGI和MODIG。结果显示,无论在AUPRC、AUROC,还是准确率Accuracy以及F1分数方面,CGMega的表现均优于其他方法(图3)。此外,准确预测癌基因通常需要大量标注信息,然而这类信息在罕见癌症研究中往往极为稀缺。因此,如何充分利用已知癌基因的相关知识对于罕见病研究至关重要。基于此,团队采用预训练和微调的策略,使CGMega在MCF7细胞系上所学到的知识可以较好地迁移到其他癌种,尤其是在有标签基因的数据少于200个时,预训练策略具有明显优势(图3)。
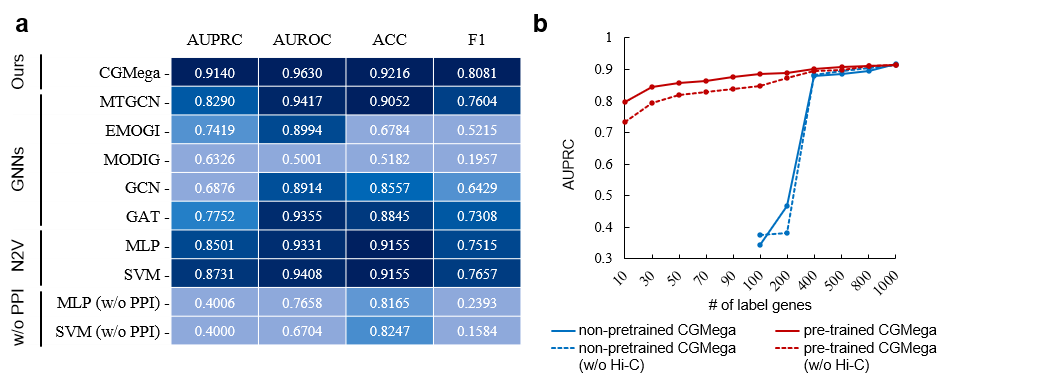
图3. 方法比较及在少标签情况下的性能
与此同时,关于多组学数据的整合方式,尤其关于染色质结构信息,团队进行了全面探索(图4)。一种策略是将染色质结构信息转化为图的拓扑结构,根据与蛋白质互作图的结合方式,可进一步分为三种方式:1)仅使用染色质结构信息;2)染色质结构图与蛋白质互作图分别经过独立的GAT编码,而后再对隐空间表示予以合并;3)染色质结构图与蛋白质互作图先融合,而后再经过统一的GAT编码。在上述三种方式中,图的节点特征相同,均由表观信息、变异信息和染色质开放信息等构成。另一种策略是仅使用蛋白质互作图作为拓扑结构,而将染色质结构信息转化为图的节点特征。根据对于Hi-C数据是否进行ICE校正、后续降维方法(如SVD、Node2Vec、LLE等)及具体维度的选择,团队对不同方法组合进行了大量测试,最终得出性能最优的整合策略是:使用蛋白质互作图作为拓扑结构,对于Hi-C数据先进行ICE校正,而后采用SVD降维到5维,然后使用这5维染色质信息和其他组学信息共同作为节点特征,对应的最优性能为AUPRC=0.9140。
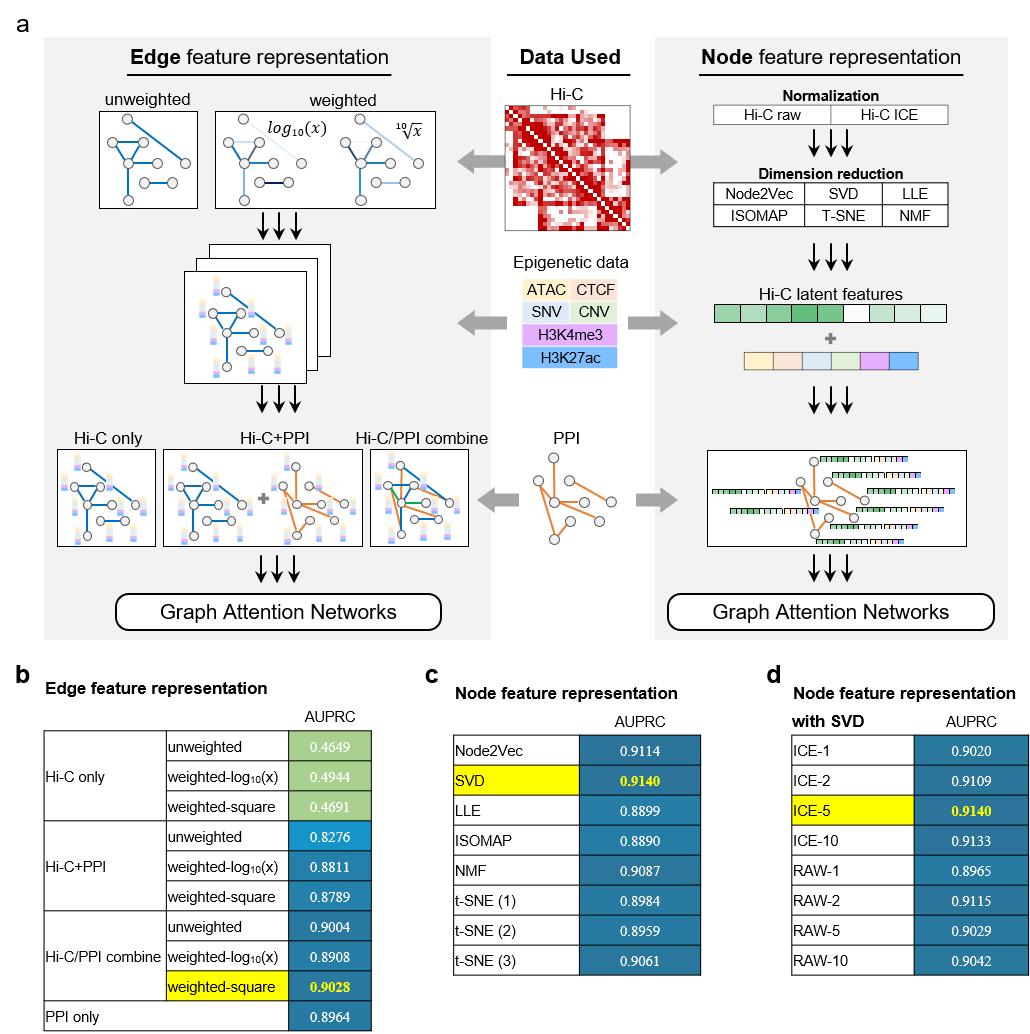
图4. 多组学信息整合策略
进一步挖掘可解释结果,团队发现对于不同的癌基因,促使CGMega做出分类决策的重要特征是不均匀分布的,由此提出代表性特征(representative features,RFs)的概念。具体表现为,有的基因如ALOX12,其代表性特征为染色质结构信息、CTCF结合情况以及组蛋白修饰;有的基因如MYB,其代表性特征为染色质结构信息与单核苷酸变异。表明染色质结构与其他组学信息具有多种联合影响癌基因的方式。此外,团队关注到ErbB家族四个基因中,HER2、HER3、ERBB4是以染色质结构信息与单核苷酸变异为代表性特征的,通过进一步整合四个基因的重要子图,发现NRG1、PPM1A、DLG2三个基因或能串联ErbB家族基因在染色质结构方面的信息交互,而以往研究则分别孤立地报导过这三个基因参与ErbB家族基因有关的肿瘤发生过程。最后,团队还进一步将CGMega运用于急性髓系白血病(AML)患者的数据,发现了不同患者之间共有和特有的驱动基因及相应模块(图5)。
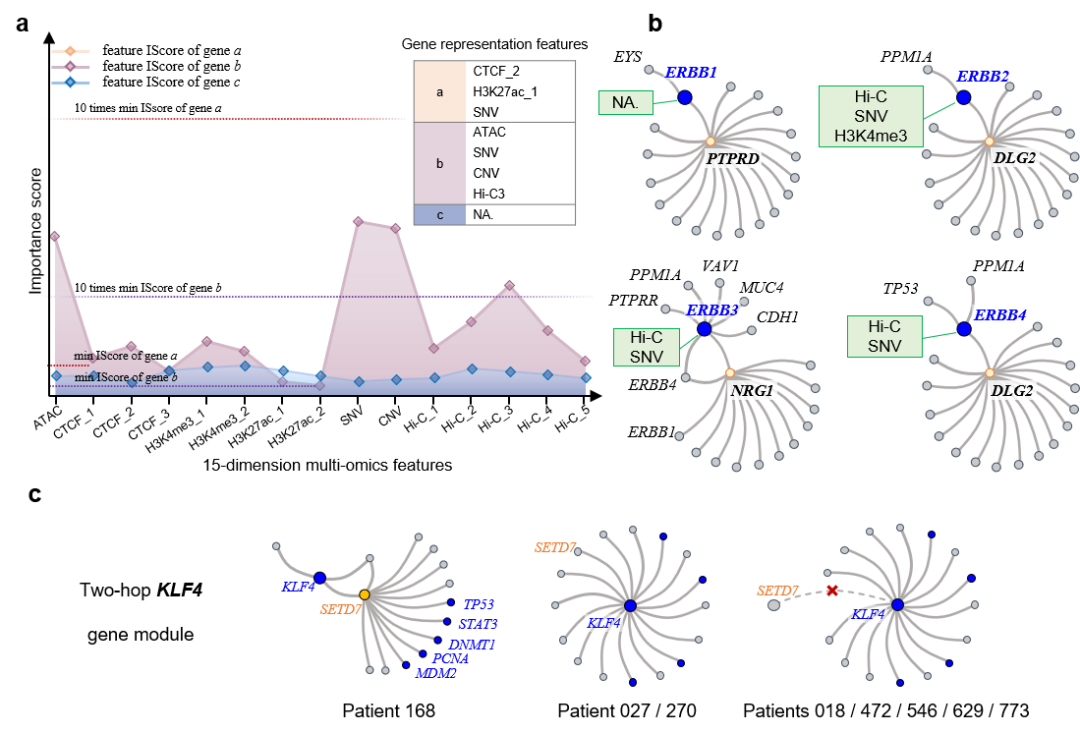
图5. CGMega可解释结果的挖掘分析
生物系统是跨越了超十个尺度的复杂信息系统,如何高效整合多模态、高维度、高噪声的组学数据,准确归纳基因组大数据背后的模式特征,精准辨识基因调控系统的关键控制元件是生物信息计算的核心挑战。CGMega计算框架为多组学数据处理和癌症基因复杂关联关系挖掘提供了新的技术途径,具有三方面技术创新性:1) CGMega通过奇异值分解(SVD)将Hi-C数据压缩成低维特征,这种方法捕捉了主要信号,避免了噪声的干扰,在癌症基因预测任务中表现出色,相比直接将Hi-C数据作为基因相互作用的度量方法更为优越,提供了更细致的基因表达特征表示,增强了模型的预测准确性。2) CGMega使用GraphTransformer代替传统的GCN架构,并增加了原始特征的残差连接,不仅可以更好地捕捉生物网络中的复杂关系,还一定程度上解决了图网络学习过程中的过平滑问题。3) 在解释模块中,CGMega引入了对称掩码矩阵约束,实现了对无向图的解释。这一创新使模型能够识别并突出显示对癌症基因预测贡献最大的相关子图结构和节点特征,确保了解释与底层图拓扑结构的一致性和连贯性。
伯晓晨研究员、杨旸教授、陈河兵副研究员为该论文的共同通讯作者。李昊副研究员、韩泽北博士、孙昱助理研究员为该论文的共同第一作者。解放军总医院血液科宁红梅主任、肿瘤内科王涛主任为该工作提供了重要指导。助理研究员任超,研究生王甫、胡朋振、许翔、白雪梅、高宇昂参与了这项工作。
伯晓晨、陈河兵团队长期从事面向多模态生物信息建模的AI方法研究,基于生成式AI技术实现了单细胞多组学数据的马赛克整合(Nat. Biotechnol. 2024),利用图神经网络有效融合了染色质可及性和三维基因组信息,实现了转录调控网络建模(Nat. Mach. Intell. 2022)和DNA双链断裂基因组特征挖掘(Genome Biol. 2023),系列分析方法为大规模组学数据分析与复杂网络关系挖掘提供了重要的原创技术。杨旸团队长期从事人工智能与科学领域的交叉研究,开发了一系列用于生物医学智能信息处理的机器学习算法模型,并获得生物实验的验证,包括基于多尺度建模方法,实现了多种类型RNA的亚细胞定位和蛋白结合位点预测(Bioinformatics 2018; BMC Genomics 2020);利用对比学习和迁移学习技术,实现了精准分子表征和体系预测(J Cheminform 2024; J. Chem. Inf. Model 2022)等。所开发算法模型为大规模复杂异质的生物医学数据的高效计算和可解释性分析处理提供了创新解决方案。
原文链接:
https://www.nature.com/articles/s41467-024-50426-6
参考文献:
1.Segal, E. et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 34, 166–176 (2003).
2.Goh, K. I. et al. The human disease network. Proc. Natl. Acad. Sci. USA 104, 8685–8690 (2007).
3.Menche, J. et al. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 347, 1257601 (2015).
4.Cheng F. et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun. Aug 2;10(1):3476 (2019)
5.Segal, E. et al. A module map showing conditional activity of expression modules in cancer. Nat. Genet. 36, 1090–1098 (2004).
6.Sadegh, S. et al. Network medicine for disease module identification and drug repurposing with the NeDRex platform. Nat. Commun. 12, 6848 (2021).
7.Wouters, J. et al. Robust gene expression programs underlie recurrent cell states and phenotype switching in melanoma. Nat. Cell Biol. 22, 986–998 (2020).
8.Sadegh, S. et al. Network medicine for disease module identification and drug repurposing with the NeDRex platform. Nat. Commun. 12, 6848 (2021).
9.Schulte-Sasse, R. et al. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 3, 513–526 (2021).
10.Zhao, W. et al. MODIG: integrating multi-omics and multi-dimensional gene network for cancer driver gene identification based on graph attention network model. Bioinformatics 38, 4901–4907 (2022).

