NEJM:IL-13单抗Lebrikizumab用于中重度特异性皮炎两项3期临床达终点(ADvocate1 和 ADvocate2研究)
时间:2023-03-16 10:00:32 热度:37.1℃ 作者:网络
特应性皮炎(AD)是一种慢性、复发性、炎症性皮肤病。由于患者常合并过敏性鼻炎、哮喘等其他特应性疾病,故被认为是一种系统性疾病。AD患者往往有剧烈瘙痒,严重影响生活质量。过去30年全球范围内AD患病率逐渐增加,发达国家儿童AD患病率达10%-20%。我国AD患病率的增加晚于西方国家,但近10年来增长迅速。我国的流行病学调查显示,学龄期青少年AD的总患病率为0.70%。
2021年8月,礼来公司宣布,来瑞组单抗(lebrikizumab) 的晚期 ADvocate 1 和 ADvocate 2 单药治疗试验也已达到其所有主要和关键次要终点。正式结果今日发表于NEJM杂志上。详细见:礼来的lebrikizumab达到了特应性皮炎研究的主要终点
两项设计相同、为期 52 周、随机、双盲、安慰剂对照、平行组、3 期试验——ADvocate1(试验 1)和 ADvocate2(试验 2)——以评估 lebrikizumab 单一疗法在治疗中的疗效和安全性 患有中度至重度特应性皮炎的成年和青少年患者。 每项试验包括两个治疗期:16 周的诱导期和 36 周的维持期。
研究人群的纳排标准如下:成人(≥18 岁)和青少年(12 至 <18 岁,体重≥40 公斤)如果患有中度至重度特应性皮炎且基线湿疹面积和严重程度指数 (EASI) 评分至少 16(范围,0 到 72,较高的值表示疾病的严重程度和范围更大),研究者的整体评估 (IGA) 评分至少为 3(范围,0 [皮肤白皙] 到 4 [严重疾病],有 描述特定时间点特应性皮炎病变整体外观的评分),受影响的体表面积至少为 10%,以及慢性特应性皮炎至少 1 年,局部治疗不充分或不可取。 EASI 评估四个身体区域的四种疾病迹象(红斑、丘疹或水肿、表皮脱落和苔藓样变)。 如果患者之前接受过 lebrikizumab、dupilumab 或 tralokinumab 治疗,则他们被排除在试验之外。
在基线访视(第 1 天)时,符合条件的患者以 2:1 的比例随机分配接受 250 mg lebrikizumab(在基线和第 2 周给予 500 mg 负荷剂量)或安慰剂,给予 每 2 周皮下注射一次。 使用电子数据采集系统进行随机化,根据地理区域(美国与欧盟与世界其他地区)、年龄组(青少年与成人)和疾病严重程度(IGA)进行分层)。 申办方、研究人员、试验现场人员和患者均不知道试验分组情况,并且在两项试验期间均保持盲法完整性。
在第 16 周之前,禁止使用局部治疗(高效、中等或低效局部糖皮质激素、局部钙调磷酸酶抑制剂或 crisaborole)或全身治疗(口服糖皮质激素、环孢菌素、dupilumab、tralokinumab 或光疗)治疗特应性皮炎,除非这种使用被认为适合作为挽救疗法。 局部治疗(例如中等效力的局部糖皮质激素)是首选的一线挽救疗法。 如果认为有必要进行全身抢救治疗,则停用 lebrikizumab。 接受救援治疗的患者有资格进入“escape”组,在完成第 16 周访视和全身救援药物清除期后,每 2 周接受一次开放标签的 lebrikizumab。
第 16 周访视结束后,符合方案规定的对 lebrikizumab 有反应标准的患者(定义为 IGA 评分为 0 或 1,且比基线减少 ≥ 2 分或 EASI 改善 75% 评分 [EASI-75 反应] 且未使用急救药物)再次随机化以接受每 2 周一次的 lebrikizumab、每 4 周一次的 lebrikizumab 或安慰剂(即,lebrikizumab 停药)。 到第 16 周没有反应的患者被分配到逃避组,接受每 2 周一次的开放标签 lebrikizumab。
主要疗效终点为第 16 周时 IGA 评分为 0 或 1,与基线相比至少减少 2 分。关键次要疗效结果包括 以下:第 16 周时的 EASI-75 响应; 第 16 周时 EASI 评分较基线(EASI-90 反应)提高 90%; 瘙痒症数字评定量表(NRS;范围,0 [无痒] 至 10 [可想象的最严重的瘙痒],用于评估患者报告的过去 24 小时内最严重的瘙痒)从基线至少减少 4 分 第 16 周; 成年患者 IGA 评分为 0 或 1,且在第 16 周时比基线降低至少 2 分; 睡眠损失量表(衡量前一晚因瘙痒干扰导致的睡眠损失程度,从 0 [“完全没有”] 到 4 [“根本无法入睡”] 至少减少 2 分 ) 从第 16 周的基线开始; IGA 评分为 0 或 1,第 4 周时比基线至少降低 2 分; 在第 4 周和第 2 周时,瘙痒症 NRS 从基线至少减少 4 分。
研究结果表明,试验 1 的患者入组时间为 2019 年 9 月 24 日至 2021 年 2 月 26 日,试验 2 的入组时间为 2019 年 10 月 29 日至 2021 年 3 月 19 日。在试验 1 中,共有 424 名患者被随机分配至 lebrikizumab 组 (283 名患者)或安慰剂组(141 名)(图 S4); 这些患者的平均(±SD)年龄为 35.5±17.3 岁,其中 55 名(13.0%)为青少年; 214 名患者 (50.5%) 为女性。 在试验 2 中,共有 427 名患者被随机分配到 lebrikizumab 组(281 名患者)或安慰剂组(146 名患者)(图 S5); 这些患者的平均年龄为 36.2±16.9 岁,其中 47 名 (11.0%) 为青少年; 211 名患者 (49.4%) 为女性。 患者的人口统计学和疾病特征在两项试验和试验组之间相似(表 1)。 总体而言,63.7% 的患者人群为白人,22.6% 为亚裔,9.9% 为黑人。
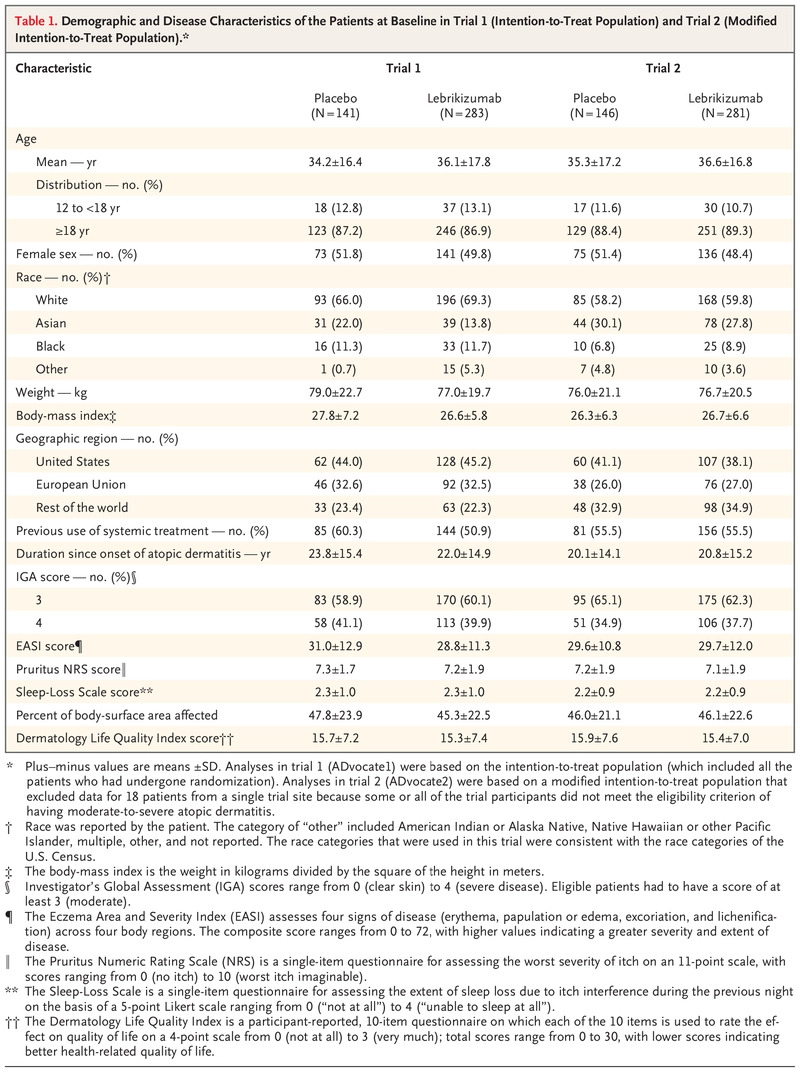
安慰剂组停止参与试验的患者比例高于 lebrikizumab 组(试验 1 中 14.9% 对 7.1%,试验 2 中 11.0% 对 7.8%)。 试验 1 中止的原因包括方案偏差(安慰剂组 3.5% 的患者和 lebrikizumab 组 2.1% 的患者)、失访(分别为 0.7% 和 1.4%)和退出 由患者(分别为 4.3% 和 1.1%)。 试验 2 中止的原因包括不良事件(安慰剂组 2.7% 的患者和 lebrikizumab 组 2.1% 的患者)、方案偏差(安慰剂组无,lebrikizumab 组 2.1%) 、患者退出(分别为 3.4% 和 1.4%)以及与 2019 年冠状病毒病大流行相关的原因(分别为 0.7% 和 1.4%)。
在这两项试验中,与安慰剂组相比,lebrikizumab 组中在第 16 周时有主要结局反应(IGA 评分为 0 或 1,与基线相比降低≥2 分)的患者百分比显着更高。在试验 1 中, lebrikizumab 组 43.1% 的患者出现主要结局反应,而安慰剂组为 12.7%(P<0.001); 在试验 2 中,相应的百分比分别为 33.2% 和 10.8% (P<0.001)(图 1A)。

关键的次要疗效结果
在两项试验中,与安慰剂组相比,lebrikizumab 组在第 16 周时出现 EASI-75 反应的患者比例更高。 在试验 1 中,lebrikizumab 组 58.8% 的患者出现 EASI-75 反应,而安慰剂组为 16.2%(P<0.001); 在试验 2 中,相应的百分比分别为 52.1% 和 18.1% (P<0.001)(图 1B)。 在试验 1 中,lebrikizumab 组中 38.3% 的患者和安慰剂组中 9.0% 的患者以及试验 2 中分别为 30.7% 和 9.5% 的患者在第 16 周观察到了 EASI-90 反应(P<0.001 对于两个比较)。

在这两项试验中,与安慰剂组相比,lebrikizumab 组中在第 16 周时瘙痒 NRS 评分较基线至少降低 4 分(两项比较均 P<0.001)且睡眠质量下降的患者百分比显着更高 -第 16 周时损失量表得分从基线至少下降 2 分(两项比较 P <0.001)。 与安慰剂组相比,lebrikizumab 组在第 4 周出现 IGA 反应(评分为 0 或 1,减少≥2 分)且瘙痒症 NRS 评分至少减少 4 分的患者百分比显着更高 第 4 周的分数。在试验 1 中,与安慰剂组相比,lebrikizumab 组中有显着更高百分比的患者在第 2 周时瘙痒症 NRS 评分至少降低 4 分 (P=0.02),但在试验 2 中没有。 与安慰剂相比,这是唯一未达到显着性阈值的次要终点。 表 2 显示了根据 FDA 多重性控制策略的所有次要结果。
安全性方面,在试验 1 中,接受 lebrikizumab 的 282 名患者中有 129 名 (45.7%) 和接受安慰剂的 141 名患者中的 73 名 (51.8%) 以及 281 名患者中的 150 名 (53.4%) 和 145 名患者中的 96 名 (53.4%) 报告了诱导期间的不良事件。 66.2%),分别在试验 2 中(表 3)。 大多数不良事件的严重程度为轻度至中度,因不良事件而终止试验的发生率很低。 接受 lebrikizumab 治疗的患者注射部位反应的发生率在试验 1 中为 1.1%,在试验 2 中为 2.1%——结果与接受安慰剂的患者相似。
最常见的不良事件(发生在接受 lebrikizumab 的患者中≥5%,并且持续报告的频率高于安慰剂组)是结膜炎(试验 1 中的患者分别为 7.4% 和 2.8%,试验 1 中的患者分别为 7.5% 和 . 试验 2) 中为 2.1%(表 3)。

结果表明,两项设计相同的 3 期试验,即 ADvocate1 和 ADvocate2,以评估 lebrikizumab 单药治疗对患有中度至重度特应性皮炎的成人和青少年的疗效和安全性均获成功。 与安慰剂相比,Lebrikizumab 治疗在第 16 周时改善了主要结局和所有次要结局,显着改善了皮肤清除率(通过 IGA 和 EASI 测量)、瘙痒(通过瘙痒 NRS 测量)、 以及瘙痒对睡眠的干扰(通过睡眠损失量表测量)。 在第 2 周和第 4 周观察到 lebrikizumab 治疗的益处关于所有次要结局,试验 2 中第 2 周的瘙痒 NRS 评分除外。这些结果显示在疾病的多个领域迅速起效,例如 作为皮肤间隙和瘙痒。 这些结果证实了 lebrikizumab 2 期试验的结果。
这些试验的结果,连同显示白细胞介素 13 和白细胞介素 13 生成细胞与特应性皮炎临床严重程度评分相关的现有证据,证实了白细胞介素 13 的核心作用 在本病的发病机制中。 与结合白细胞介素 4 和白细胞介素 13 受体复合物共有的白细胞介素 4Rα 受体亚基的 dupilumab 相比,lebrikizumab 和 tralokinumab 直接结合 interleukin-13。与 tralokinumab 相比,lebrikizumab 不干扰 interleukin-13Rα2,它是人体消除过量 interleukin-13的自然机制的一部分。 体外研究表明,与 tralokinumab 相比,lebrikizumab 具有更高的结合亲和力和更慢的解离速率。
这些试验中的诱导期(16 周)限制了对长期治疗的疗效和安全性的评估。 来自这两项临床试验维持期和长期扩展研究(ADjoin;NCT04392154。在新标签中打开)的更多信息可能会有用。 此外,关于单一疗法的数据可能并不总能转化为现实生活中的临床环境。 进行了一项单独的临床试验,以评估 lebrikizumab 联合外用糖皮质激素治疗中度至重度特应性皮炎患者的安全性和有效性。
在两项随机、安慰剂对照的 3 期试验中,lebrikizumab 治疗显着改善了成人和青少年中度至重度特应性皮炎的体征和症状。
事实上,有关这两项研究的长期结果也有公布,在 ADvocate 1 研究中,治疗一年后,79%每4周接受lebrikizumab治疗的患者和 79% 每2周接受lebrikizumab治疗的患者保持75% 或更高的皮肤改善(EASI-75)。在ADvocate 2研究中,治疗一年后,85%每4周接受lebrikizumab的患者和 79% 每2周接受lebrikizumab的患者维持 75% 或更高的皮肤改善(EASI-75)。
原始出处:
Two Phase 3 Trials of Lebrikizumab for Moderate-to-Severe Atopic Dermatitis,NEJM,March 15, 2023
DOI: 10.1056/NEJMoa2206714

