梅斯盘点:FDA在2022年度批准的创新药(上)
时间:2023-01-03 15:01:38 热度:37.1℃ 作者:网络
截至2022年12月30日,美国FDA的药物评估和研究中心(CDER)已经批准了37款创新药。FDA的生物制品评估和研究中心(CBER)也批准了至少15项生物制品许可申请(BLA)。虽然与往年相比,今年FDA批准的新药数目有所下降,但是创新的步伐并没有减缓。在FDA今年批准的新药中,新分子疗法的占比达到了历史新高。“First-in-class”疗法的比例在过去八年中也位居第一。FDA药物评估和研究中心批准了至少19款“first-in-class”疗法,占获批新药总数的56%。
新分子疗法的定义包括双特异性抗体/蛋白、RNAi疗法、反义寡核苷酸疗法(ASO)、基因疗法(含基于细胞的基因疗法)、mRNA疗法或疫苗、溶瘤病毒、微生物组疗法和抗体偶联药物(ADC)。
FDA已经批准了15款新分子疗法,新分子疗法无论是获批的数目还是在FDA批准中的占比都达到近10年来的最高,显示了新治疗模式蓬勃发展的趋势。这15款新分子疗法中,包含了5款涉及基因改造的细胞和基因疗法,4款双特异性抗体/蛋白疗法,1款RNAi疗法,1款mRNA疫苗,1款微生物组疗法和1款抗体偶联药物。
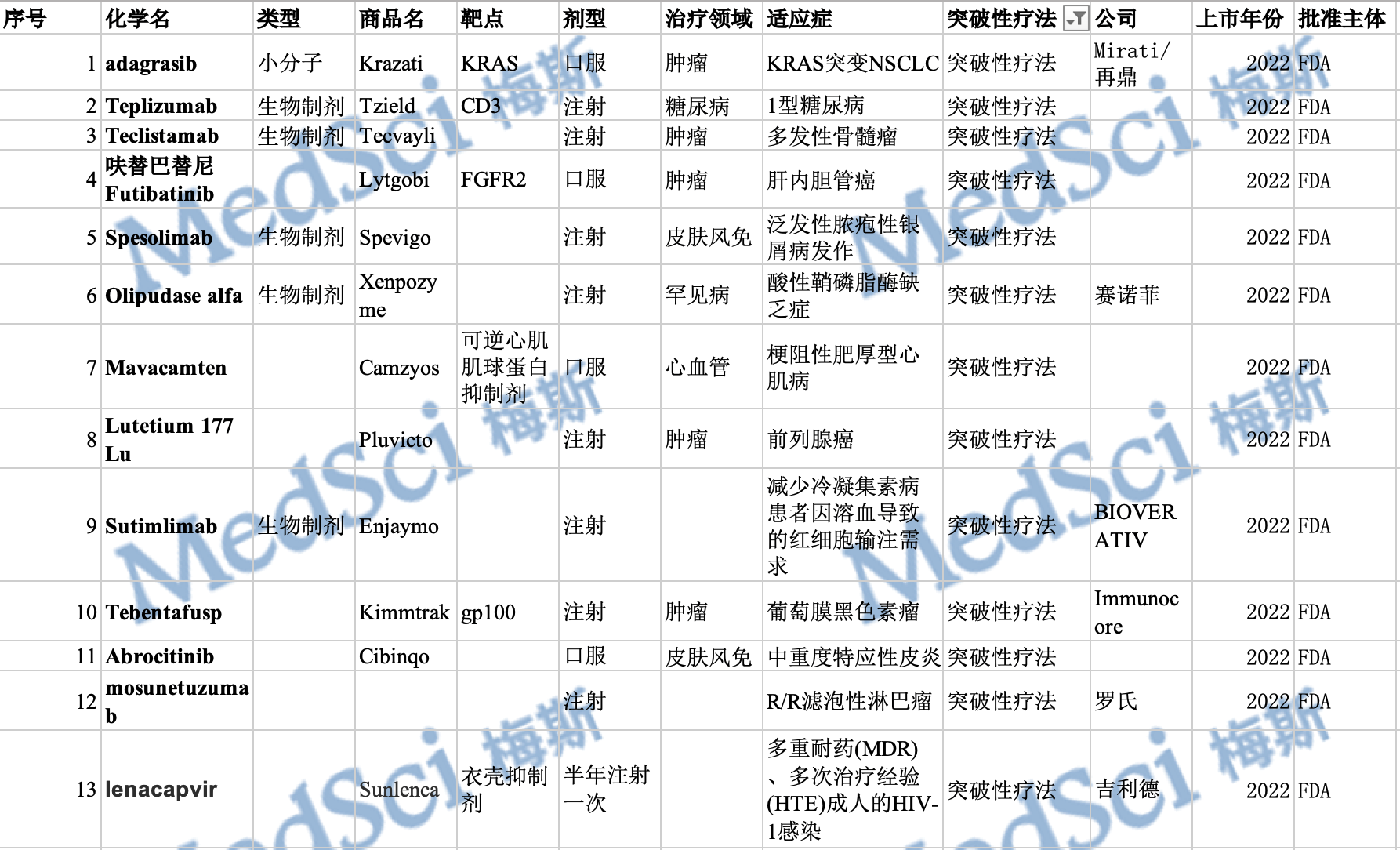
图1 2022年度获突破性疗法认定的药物
DARIDOREXANT(商品名:QUVIVIQ)
1月7日,FDA批准了IDORSIA公司的DARIDOREXANT,用于成人失眠症治疗。本品是一种食欲素受体阻滞剂,与Suvorexant作用机制相同。FDA批准本品上市是基于两项三期临床试验的结果,在两项临床试验中,1854名受试者分别接受了本品50mg,本品25mg或安慰剂治疗,主要终点为治疗后1个月、3个月的入睡后觉醒时间(WASO)与睡眠潜伏期(LPS)相比基线的变化。结果显示,在治疗1个月后,50mg组、安慰剂组合安慰剂组的WASO分别相比基线缩短29、18和6分钟,LPS分别基线缩短31、28和20分钟,而治疗3个月后,三组的WASO分别相比基线缩短29、23和11分钟,LPS则分别相比基线缩短35、31和23分钟。两项临床试验说明,经过本品治疗,患者睡眠质量得到显著改善。详细见:2022年1月,FDA批准两款创新药,分别主治失眠和特异性皮炎
ABROCITINIB (商品名:CIBINQO)
1月14日,FDA批准了辉瑞公司的ABROCITINIB,用于难治性的中重度特应性皮炎治疗。本品是一种可逆性的JAK1抑制剂,三项临床试验评估了本品的安全有效性。三项临床试验共计纳入了1615名患者,主要终点均为12周的研究者整体评分(IGA)为0分或1分的患者比例和湿疹面积及严重程度指数相比基线改善75%以上患者的比例(EASI-75)。试验一和实验二为单药治疗,试验三为联合用药治疗。12周治疗结果显示,试验一中,本品200mg、100mg和安慰剂组IGA为0或1分的患者比例为44%,24%和8%,而试验二分别为38%,28%和9%,试验三则分别为47%,36%和14%。EASI方面,试验一中三组患者分别有62%,40%和12%的患者达到EASI-75标准,而试验二分别为61%、44%和10%,试验三则分别为68%,58%和27%。详细见:2022年1月,FDA批准两款创新药,分别主治失眠和特异性皮炎
Tebentafusp(商品名Kimmtrak),全球首款TCR双抗疗法
1月25日,FDA批准了IMMUNOCORE公司T细胞受体(TCR)疗法TEBENTAFUSP,用于HLA-A*02:01阳性,且不可切除或转移性葡萄膜黑色素瘤治疗。本品是一种双特异性 gp100 肽-HLA 介导的CD3 T细胞接合剂,TCR臂可与黑色素瘤细胞的细胞表面的gp100 肽结合,进而发挥治疗作用。在一项临床试验中,378名符合条件的患者按2:1分组,分别接受本品或标准疗法(PD-1,CALT4或化疗)治疗,中位总生存期分别为21.7个月 vs 16.0个月,中位无进展生存期分别为3.3个月 vs 2.9个月,可观应答率为9.1% vs 4.8%。临床试验结果说明,相比现有的疗法,本品可显著降低患者的死亡风险。详细见:NEJM:转移性葡萄膜黑色素瘤采用Tebentafusp治疗的总生存获益
FARICIMAB(商品名:VABYSMO)
1月28日,基因泰克公司的FARICIMAB喜获FDA批准,用于年龄相关的黄斑病变或糖尿病黄斑病变治疗。本品是一种血管表皮细胞生长因子和血管生成素双重抑制剂,两项临床试验证实了本品对年龄相关的黄斑病变的安全有效性。两项试验共计有1329名患者入组,经过长达2年的治疗和随访,试验结果本品的疗效非劣于阿柏西普。糖尿病黄斑病变方面,该公司同样开展了两项临床试验,两项试验共计有1891名患者入组,两项临床试验的结果同样证实,本品的疗效非劣性于阿柏西普。详细见:Lancet:双特异性抗体faricimab在湿性或新生血管性年龄相关性黄斑变性(AMD)和糖尿病性黄斑水肿(DME)1年的随访结果
SUTIMLIMAB(商品名:ENJAYMO)
2月4日,FDA批准了BIOVERATIV医疗公司的SUTIMLIMAB,用于冷凝集素病(CAD)患者治疗,以降低患者因溶血而对红细胞输注的依赖。CAD是冷抗体型自身免疫性溶血性贫血,是免疫球蛋白M引起的免疫性疾病,而本品是一种经典补体抑制剂,可与补体C1结合,抑制经典补体途径。在一项开放标签,单臂设计的临床试验中,24名患者接受了本品治疗,其中13名达到应答标准(血红蛋白浓度超过12 g/dL或血红蛋白浓度相比基线增加2 g/dL以上,在治疗后第5-26周无红细胞输注、无需接受标准疗法治疗)。其中9名患者血红蛋白浓度超过了12 g/dL,15名相比基线提升了2 g/dL,在5-26周间,17名患者未接受红细胞输注,22名患者未接受CAD标准方案治疗。CAD是一种极其罕见的疾病,患病率约1/10万-2/10万,本品是首个针对性的疗法。详细见:NEJM:sutimlimab治疗冷凝集素病效果喜人
MITAPIVAT(商品名:PYRUKYND)
2月17日,FDA批准了AGIOS制药的MITAPIVAT,用于丙酮酸激酶(PK)缺乏所引起的溶血性贫血治疗。PK缺乏症是一种由丙酮酸激酶基因异常所致的溶血性贫血,而本品是一种PK激动剂,FDA批准本品是基于两项临床试验的研究结果。其中一项是针对未定期输血的患者(试验开始前52周输血不超过4次,3个月内未接受输血),共有80名患者加入到临床试验,试验的主要终点为第16、20和24周时,血红蛋白浓度相比基线增加1.5g/dL的患者比例。其结果显示,本品治疗的40名患者,16%的患者出现了应答,而安慰剂组治疗的患者,应答率为0%。另一项试验是针对定期输血患者设计的开放性试验(试验开始前的52周内至少有6次输血),共有27名患者参与了试验,试验的主要终点为在一定时期内,患者的红细胞输注量相比历史下降33%以上。结果显示,33%的患者达到了应答标准,其中22%的患者未输血。详细见:NEJM:Mitapivat治疗丙酮酸激酶缺乏症的疗效(ACTIVATE试验)、FDA批准Pyrukynd治疗成人溶血性贫血
VONJO(商品名:PACRITINIB)
2月28日,FDA加速批准了CTI生物制药公司的PACRITINIB,用于血小板计数低于50×109/L的中度或高危原发性或继发性的骨髓纤维化。本品是一种JAK2和FLT3双重抑制剂。Pacritinib对野生型JAK2、突变型 JAK2(V617F)和FLT3都具有活性,这有助于与造血和免疫相关的细胞因子和生长因子的信号传导。骨髓纤维化通常与失调的 JAK2 信号传导有关。与同家族的JAK1、JAK3 和 TYK2 相比,pacritinib对JAK2 具有更高的抑制活性。Pacritinib 也表现出对其它激酶家族(如 CSF1R 和 IRAK1)的抑制活性,但其临床相关性尚有待研究。在一项311名患者参与的临床试验中,本品的安全有效性得到了初步的确证。入组患者按1:1:1的比列分别接受本品400mg每日一次、本品200mg每日2次和现有的最佳方案治疗。在该试验中,每日一次400mg未确定安全性,也未获得批准,而在每日两次200mg,且基线血小板计数低于 50 × 10 9的患者队列中获得了有效性数据。31名患者接受本品每日两次200mg治疗24周,29%的患者脾脏体积相比基线缩小了35%以上,而接受标准疗法的32名患者中,仅有3.1%达到这一标准。详细见:FDA批准骨髓纤维化新药pacritinib
CARVYKTI(西达基奥仑赛)
2022年2月28日,第一款完全国产的CAR-T细胞疗法获FDA批准上市!由南京传奇生物研发的CARVYKTI(西达基奥仑赛)的国产CAR-T细胞疗法获FDA批准上市,用于治疗复发难治性多发性骨髓瘤。详细见:FDA批准传奇生物首款CAR-T细胞治疗产品CARVYKTI®(西达基奥仑赛)上市
GANAXOLONE(商品名:ZTALMY)
3月18日,FDA批准了MARINUS公司的GANAXOLONE,用于细胞周期蛋白依赖性激酶样-5 (CDKL5)缺乏症相关的癫痫治疗。CDKL5缺乏症是一种严重而罕见的遗传性癫痫,本品的作用机制尚未完全清楚。FDA批准该产品上市,是基于一项有101名患者参与的临床试验,患者按1:1的比例进行分组后,分别接受本品或安慰剂治疗,主要终点是28天的癫痫发作频率相比基线值的变化。经过治疗,安慰剂组和本品治疗组的癫痫发作频率分别相比基线值下降7%和31%,达到了统计学上的显著性差异。详细见:FDA批准GABA受体别构调节剂ganaxolone上市、Lancet Neurol:Ganaxolone治疗CDKL5缺乏症的疗效
NIVOLUMAB和RELATLIMAB(商品名:OPDUALAG)
同在3月18日,FDA也批准了百时美施贵宝公司的新型免疫疗法OPDUALAG,用于无法手术或转移性黑色素瘤治疗。本品由PD-1抗体NIVOLUMAB和LAG-3单抗RELATLIMAB组成,RELATLIMAB是一种全新的免疫检查点抑制剂。在一项临床试验中,714名三级或四级无法手术或转移性的黑色素瘤患者,分别随机接受本品(n=355)或NIVOLUMAB单独治疗(n=359),结果显示,中位无进展生存期分别为10.1个月 vs 4.6个月,中位总生存期分别为NR vs 34.1个月,可观应答率分别为43% vs 33%。相比单药疗法,本品的疾病控制率大幅提高,死亡风险下降了25%。详细:FDA批准LAG-3抗体Relatlimab联合Nivolumab用于不可切除或转移性黑色素瘤的儿童和成人患者
LUTETIUM LU-177 VIPIVOTIDE TETRAXETAN(商品名:PLUVICTO)
3月23日,诺华的新型放射型药物喜获FDA批准,用于前列腺特异性膜抗原(PSMA)阳性的去势抵抗的转移性前列腺癌。PLUVICTO是诺华公司(Novartis)研发的“first-in-class“放射性药物,用于治疗前列腺特异性膜抗原(PSMA)阳性转移性去势抵抗性前列腺癌(mCRPC)。PSMA是一种肿瘤相关抗原和 II 型跨膜蛋白,多在数恶性前列腺癌细胞中高表达。Pluvicto由 PSMA-617(一种 靶向PSMA的配体)与放射性同位素镥177Lu耦合。通过静脉注射Pluvicto后,PSMA-617靶向并结合到表达PSMA的肿瘤细胞表面,并在受体循环过程中进入细胞内,随后177Lu通过衰变释放的β粒子破坏肿瘤细胞。在一项临床试验中,患者按2:1的比例分组,分别接受本品联合最佳支持治疗(BSoC)或BSoC单独治疗,结果显示接受联合治疗的551名患者,中位总生存期为15.3个月,而接受BSoC单独治疗的280名患者,中位总生存期仅为11.3个月,两组患者的客观缓解率分别为30% vs 2%,达到了预设的主要终点。详细见:FDA批准放射性配体疗法Pluvicto治疗前列腺癌
OTESECONAZOLE(商品名:VIVJOA)
4月26日,FDA批准了MYCOVIA制药的OTESECONAZOLE,用于外阴道念珠菌病复发预防。本品是一种新型抗真菌药,体外试验证实对多种念珠菌均有活性,FDA批准本品上市,是基于三项临床试验的研究结果。其中试验一和试验二为安慰剂对照的随机试验,试验重点皆为48周内,念珠菌培养确证的急性发作((念珠菌属真菌培养阳性,临床体征和症状评分为≥3))次数≥1次的患者比例。结果显示,试验一中,本品治疗组和安慰剂组患者的急性发作比例分别为6.7%和42.8%,试验二中,两组患者的急性发作比例分别为3.9%和39.4%,均达到了预设的终点。详细见:FDA批准oteseconazole(奥特康唑)用于复发性外阴阴道假丝酵母菌病治疗
MAVACAMTEN(商品名:CAMZYOS)
4月28日,FDA批准了百时美施贵宝公司的MAVACAMTEN,用于梗阻性肥厚型心肌病治疗,以改善患者的心脏功能和症状。本品是一种选择性的可逆心肌肌球蛋白抑制剂,FDA批准本品上市是基于一项三期试验的研究结果。251名纽约心脏病协会(NYHA)分组为II-III级的梗阻性肥厚型心肌病患者分别接受本品或安慰剂治疗,试验终点为30周时评估心脏功能,混合静脉血氧张力改善≥1.5 mL/kg/min外加NYHA分组改善1级以上的患者比例或混合静脉血氧张力改善≥3.0 mL/kg/min外加NYHA分组无恶化的患者比例。结果显示,接受本品治疗的123名患者中,37%的患者达到了试验终点,而接受安慰剂治疗的128名患者中,仅有22%的患者达到了试验终点。试验结果说明,本品相比安慰剂,可显著性改善患者的心脏功能。详细见:联拓生物合作伙伴百时美施贵宝公司的Camzyos (mavacamten) 获美国FDA批准
阿莫西林、克拉霉素和沃诺拉赞(商品名:VOQUEZNA TRIPLE PAK)
5月3日,FDA批准了PHATHOM 公司的VOQUEZNA TRIPLE PAK,用于幽门螺旋杆菌感染治疗。本品含有β-内酰胺类抗生素阿莫西林,大环内酯类抗生素克拉霉素和新型抗酸药沃诺拉赞,FDA批准本品上市是基于一项三期临床试验的研究结果。入组患者按1:1:1的比例,分别接受沃诺拉赞20mg每日二次+阿莫西林1000mg每日二次+克拉霉素500mg每日二次(沃诺拉赞三联治疗组),沃诺拉赞20mg每日二次+阿莫西林1000mg每日三次(沃诺拉赞二联治疗组),或兰索拉唑30mg每日两次+阿莫西林1000mg每日二次+克拉霉素500mg每日二次(兰索拉唑赞三联治疗组)治疗,治疗27天后,幽门螺旋杆菌检测阴性为治疗成功。结果显示,沃诺拉赞三联治疗组治疗成功率为80.8%,沃诺拉赞二联治疗组治疗成功率为77.2%,而兰索拉唑赞三联治疗组仅为68.5%。
TIRZEPATIDE(商品名:MOUNJARO),首个GLP-1和GIP双靶点激动剂
5月13日,FDA批准了礼来公司的替扎罗(Mounjaro, tizepatide),在饮食控制和运动的基础上,用以控制二型糖尿病患者的血糖。
本品是一种GLP-1和GIP双靶点激动剂,为了获得FDA的批准,礼来公司已经开展了多项临床试验研究。单药治疗方面,经过本品不同剂量的40周治疗,本品5mg、10mg和15mg治疗组的糖化血红蛋白分别相比基线下降了1.8%、1.7%和1.7%,而安慰剂组仅下降了0.1%,三个治疗组患者的平均体重分别相比基线下降了6.3kg、7.0kg和7.8kg,而安慰剂组仅下降了1.0kg。在二甲双胍为背景治疗的基础上添加本品治疗方面,本品5mg、10mg和15mg治疗组的糖化血红蛋白分别相比基线下降了2.0%、2.2%和2.3%,而索马鲁肽治疗组(对照)则仅下降了1.9%,体重方面,本品三个治疗组分别相比基线下降7.6kg、9.3kg和11.2kg,而索马鲁肽治疗组则仅下降了5.7kg。在基础胰岛素的背景治疗下,添加本品治疗方面,本品5mg、10mg和15mg治疗组的糖化血红蛋白分别相比基线下降了2.1%、2.4%和2.3%,而安慰剂组仅下降了0.9%,体重方面,本品三个治疗组分别相比基线下降5.4kg、7.5kg和8.8kg,而安慰剂组则上涨了1.6kg。Lilly产品在肥胖症方面也表现出令人印象深刻的功效。在最高剂量下,该药物比诺和诺德(Novo Nordisk)的GLP1受体激动剂Wegovy(semaglutide)更能减轻体重。再加上该药物具有更好的耐受性,预测上市后仅6年,年销售额约可达80亿美元。事实上,一些较为乐观的卖方分析师预测糖尿病和肥胖症的最高销售额将达到200亿美元——这一数字将使Mounjaro成为商业上最成功的糖尿病药物之一。详细见:NEJM:Tirzepatide一周一次治疗严重肥胖,效果极佳!(SURMOUNT-1研究)、NEJM:Tirzepatide头对头III期研究成功 降糖减重效果优于索马鲁肽
TAPINAROF(商品名:VTAMA)
5月23日,FDA批准了DERMAVANT公司的TAPINAROF,用于斑块银屑病治疗。本品是一种芳香烃受体激动剂,两项安慰剂对照的临床试验证实了本品的安全有效性。在两项试验中,患者均按2:1的比例分组,分别接受DERMAVANT乳膏和安慰剂治疗,试验终点均为12周时PGA评分为0分(斑块清除)和1分(几乎清除)的患者比例。结果显示,两项试验中的本品治疗组分别有36%和40%的患者达到了治疗终点,而安慰剂组则分别只有6%和6%。详细见:NEJM:Tapinarof乳膏局部治疗银屑病III期临床获得成功、25年来首次!FDA批准银屑病外用新药上市——本维莫德乳膏
VUTRISIRAN(商品名:AMVUTTRA)
6月13日,FDA批准了ALNYLAM公司的VUTRISIRAN,用于遗传性转甲状腺素淀粉样变所致的多发性神经病治疗,本品是一种由GalNAc递送的小干扰RNA,它是其开创性siRNA药物Onpattro (patisiran)的后续产品。Amvuttra被批准用于治疗遗传性转甲状腺素介导(hATTR)淀粉样变性患者的多发性神经病,这是一种损害神经纤维的罕见疾病。尽管与Onpattro相比,Amvuttra表现出显著的剂量和便利性优势——每三个月一次皮下注射,而不是每三周一次输注——但目前尚不清楚接受Onpattro治疗的患者是否会转换使用Amvuttra,一些人推测Amvuttra的最初使用可能用于新患者。FDA批准本品上市是基于一项开放标签的临床试验结果。入组患者按3:1的比例分别接受本品或patisiran治疗,主要终点为9个月时患者调整神经病变缺损评分(mNIS+7)相比基线值的变化,结果显示,本品治疗组相比基线下降了2.2分,而安慰剂组则上升了14.8分。相比使用脂质复合物递送的patisiran,本品虽然疗效无显著性提升,但只需要三个月一次皮下注射,患者的顺应性得到大幅提升。详细见:FDA批准vutrisiran治疗成人遗传性转甲状腺素蛋白淀粉样变性伴多发性神经病(hATTR-PN),这是第5款siRNA药物
下部分:梅斯盘点:FDA在2022年度批准的创新药(下)
