新药研发利器PROTAC技术全面解析:原理,优势,挑战,机遇与下一代PROTAC技术
时间:2021-08-29 22:02:59 热度:37.1℃ 作者:网络
在过去的几十年里,小分子靶向药在延长患者生存期和改善患者生存质量上已经取得了巨大的成功。但是传统的小分子靶向药只能靶向大约 20%的蛋白,仍有大量的无酶功能的蛋白质,如转录因子、支架蛋白,或者与内源性的配体有很强结合力的蛋白(如 Ras等), 很难被传统小分子药物靶向。这类“不可成药”靶点的药物研发极具挑战,而基于蛋白降解的 PROTAC (Proteolysis-Targeting Chimera)技术在靶向不可成药靶点方面展现出了巨大的潜力。在细胞治疗、抗体药和基因编辑等新兴技术风头正盛的今天,PROTAC技术有望带领小分子药物重回巅峰。在此,我们将为大家介绍PROTAC的技术原理、优势。同时,我们将回顾在药物发现过程中有关蛋白靶向降解的经验教训,并讨论PROTAC在癌症治疗中面临的挑战和机遇。
▉ PROTAC概述
PROTAC (Proteolysis Targeting Chimeras,蛋白降解靶向联合体)的概念最早是在2001年由Crews等人提出,能够利用机体内天然存在的蛋白清理系统,降低蛋白水平而非抑制蛋白的功能,发挥治疗疾病的目的。PROTAC技术经过20年的不断完善和发展,俨然成为目前新药研发领域最火热的技术之一,受到科研院所、药企和投资机构的青睐。
 Eur J Med Chem 2020, 208, 112781.
Eur J Med Chem 2020, 208, 112781.
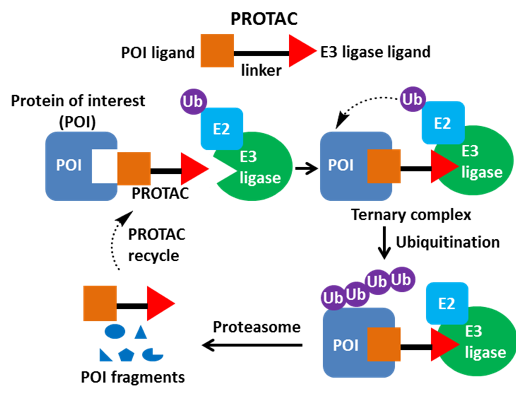
PROTAC是一种异双功能分子,分子的一端连接结合靶蛋白的配体,一端连接E3连接酶的配体,中间通过合适的Linker相连。PROTAC降解靶蛋白是通过泛素蛋白酶体系统(UPS)实现的,其大致过程如下,PROTAC分子结合靶蛋白(POI)和E3连接酶,形成三元复合物,给靶蛋白打上泛素化的标签,泛素化的蛋白被细胞内的蛋白酶体26S识别并降解。
▉ PROTAC的优势 (1)作用范围更广、活性更高、可靶向“不可成药”靶点传统的小分子和抗体等都是通过“占据驱动”的作用模式抑制靶蛋白的功能发挥治疗疾病的作用,这种作用模式需要抑制剂或单抗具备较高的浓度才能够占据靶点的活性位点,阻断下游信号通路的转导。而PROTAC是“事件驱动”,不是影响蛋白的功能,而是介导致病靶蛋白被降解。只要PROTAC介导三元复合物的形成并给靶蛋白打上泛素化的标签,理论上是可以循环反复使用的,因此催化剂量即可发挥作用。而且PROTAC对于没有活性位点的蛋白,如支架蛋白等,只要能够产生结合作用就可以诱导相关蛋白被降解,可以大大提高靶点的范围。
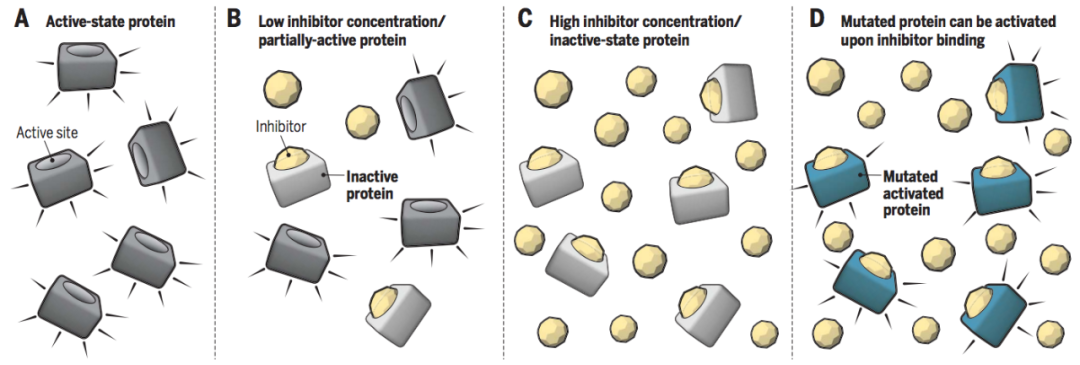
传统小分子抑制剂的“占据驱动” 。Science. 2017, 355(6330): 1163-1167
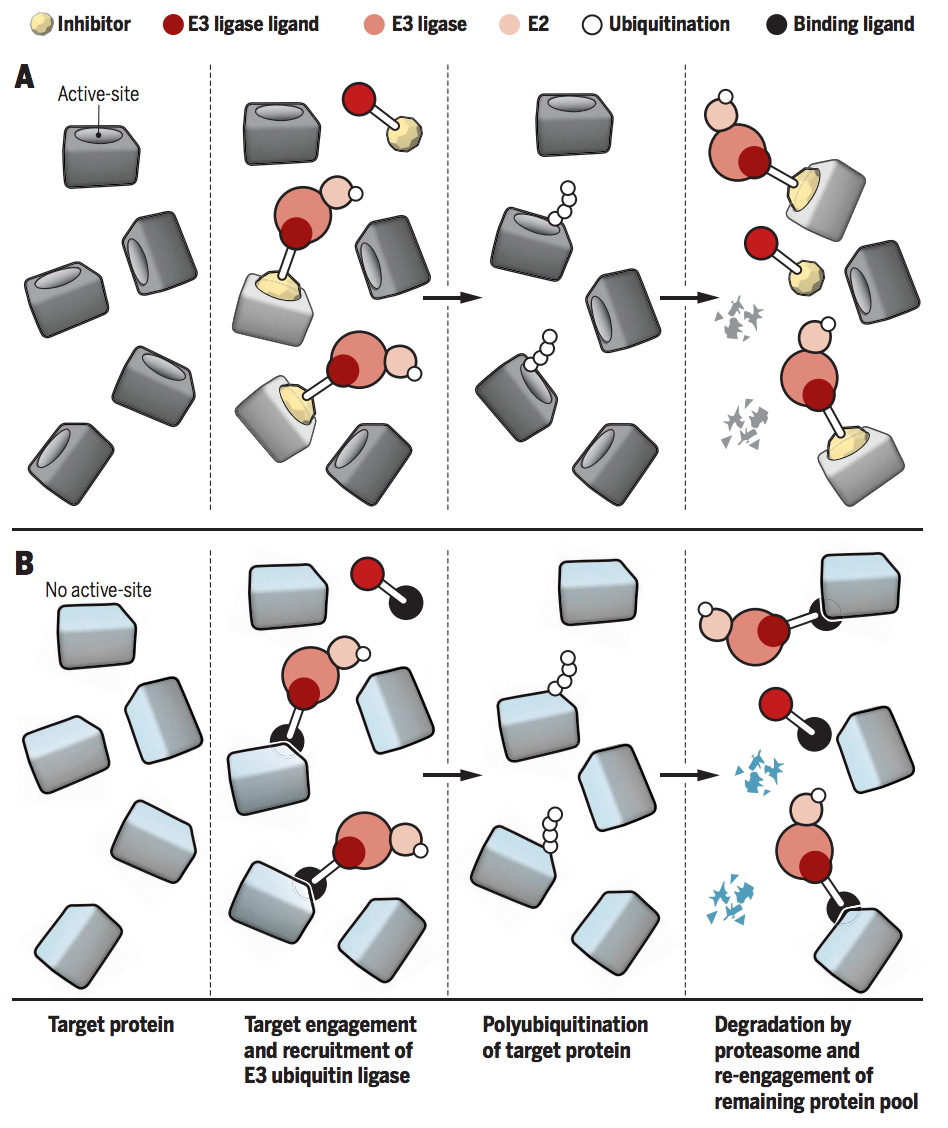
PROTAC的“事件驱动”。Science. 2017, 355(6330): 1163-1167.
据不完全统计,目前已经有超过100种蛋白被成功降解。这些靶点包括(1)激酶类,如RIPK2、BCR-ABL、EGFR、HER2、c-Met、TBK1、CDK2/4/6/9、ALK、Akt、CK2、ERK1/2、FLT3、PI3K、BTK、Fak等;(2)BET蛋白,如BRD2/4/6/9;(3)核受体,如AR、ER等;(4)其他蛋白,如MetAp-2、Bcl-xL、Sirt2、HDAC6、Pirin、SMAD3、ARNT、PCAF/GCN5、Tau、FRS2等。这其中就包括“不可成药靶点”,如转录因子调节蛋白pirin、表观遗传相关蛋白PCAF/GCN5等。另外,根据Nature 3月份的一项报告显示,到2021年年底,将至少有15款蛋白降解剂(包含PROTAC和分子胶)进入临床试验。
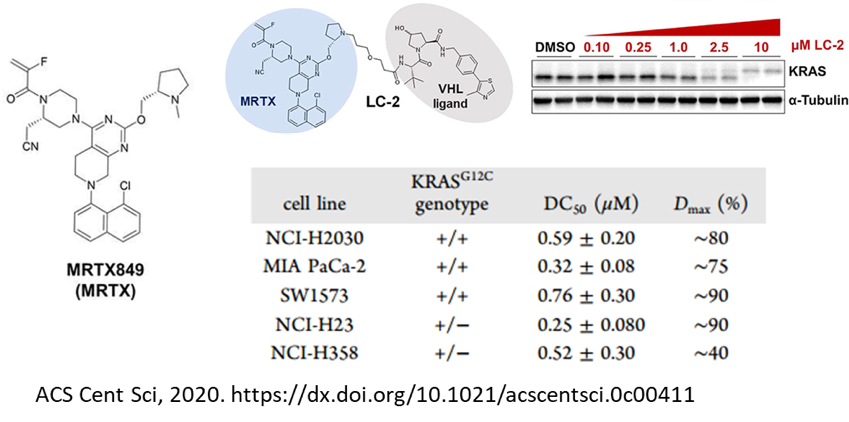
说到不可成药靶点,RAS(KRAS、HRAS和NRAS)肯定是绕不要过去的。作为癌症中最常见的突变基因,RAS是肺癌、结直肠癌和胰腺癌的重要驱动因素。被发现和研究40多年来,一直没有针对该靶点的药物上市。终于在今年5月份,Amgen公司开发的KRAS-G12C不可逆抑制剂AMG510(Sotorasib)成功上市,总算是终结了该靶点的“不可成药性”。虽然KRAS的不可成药性被小分子抑制剂终结了,但是降解剂在该靶点仍然大有可为。针对KRAS-G12C突变,Crews等人在KRAS抑制剂AMG510和MRTX849的基础上,设计并合成了相关的PROTAC,活性测定发现了具有良好降解活性的降解剂,这或许可以为进一步攻克RAS提供新的解决方案。可以预见,利用PROTAC技术攻克的可能不只是G12C,对其他突变或许也能有所作为。
 (2)提高选择性、活性和安全性
(2)提高选择性、活性和安全性
与传统小分子抑制剂相比,PROTAC在某些靶点上可实现小分子难以实现的选择性。例如,多靶点酪氨酸激酶抑制剂Foretinib可以结合130多种激酶,Crews等人将其作为结合靶蛋白的配体,分别连接E3连接酶VHL(von Hippel-Lindau)和CRBN(cereblon)的配体得到相应的PROTAC,结果显示连接VHL和CRBN的PROTAC只能分别降解36和62种蛋白,而只有12种蛋白才能被这两种PROTAC降解。另外一项Gray课题组的研究也证明了PROTAC可以实现靶点的选择性。2,4-二氨基嘧啶骨架是激酶抑制剂的常见骨架,EGFR、ALK、CDK、Jak等激酶的抑制剂都有用到其作为母核。但是该骨架形成的药物分子大多都比较“脏”,也就是靶点选择性比较差,经常对其他很多激酶都有很强的抑制活性,开发过程中off target的脱靶效应常常成为毒副作用的主要来源,影响新药开发的成功率。Gray课题组基于该骨架合成的PROTAC虽然能够结合190多种激酶,但是细胞实验中只能降解12和22种激酶,大大提高了靶点的选择性。由此可见,合理的药物设计加之反复迭代优化,选择性更高、活性更好、安全性更佳的PROTAC分子很有可能被发现。
PROTAC技术不仅可以实现小分子抑制剂难以做到的选择性,在提高活性方面也具有非常显著的优势。以PROTAC技术应用最早的靶点BET(BRD2/3/4)为例,秦冲等人发现的QCA570在细胞抗增殖活性方面表现出了明显优于JQ1等抑制剂的能力,JQ1等抑制剂的活性大都在纳摩尔级水平,而QCA570的活性提高了三个数量级,达到了惊人的皮摩尔级水平。基于出色的体外细胞抗增殖活性,降解剂在体内也表现出了强效抗肿瘤活性,而且呈现出给药剂量低和给药频次也低的特点。这说明在类似BET等靶点中,PROTAC具有明显的优势。但是一直未见有针对该靶点的降解剂进入临床,具体原因不明。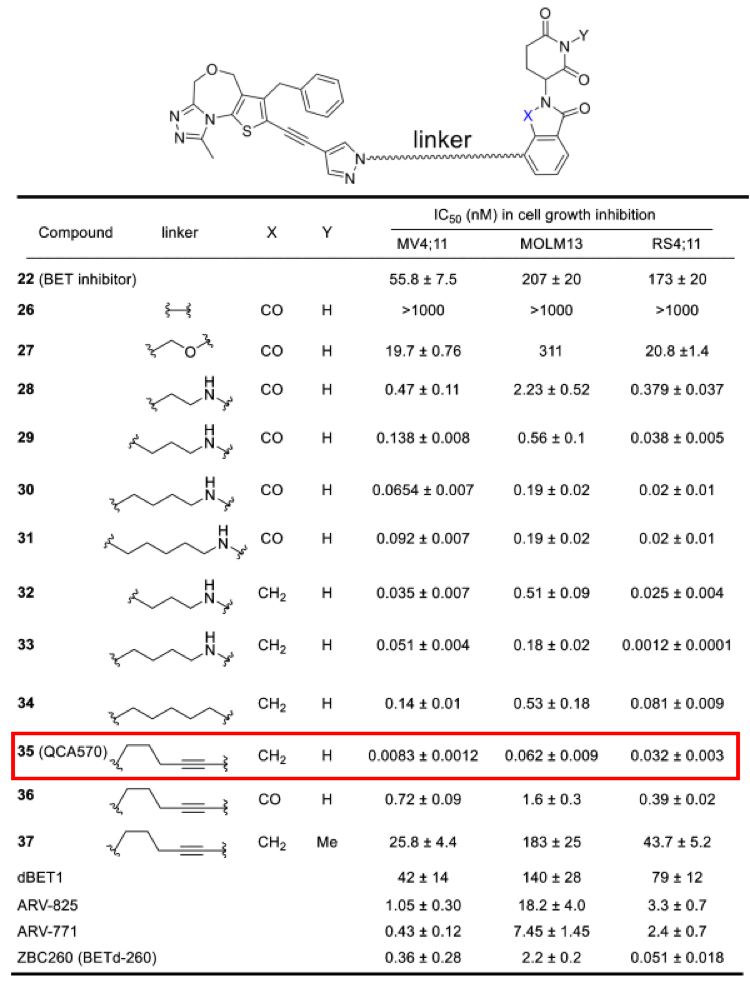
J Med Chem 2018, 61, 6685-6704.
(3)克服药物的耐药性
小分子抑制剂或拮抗剂在临床用药过程中,不可避免的都会发生获得性耐药。比如EGFR-T790M和C797S耐药等。虽然可以通过开发新一代的抑制剂,如第三代和第四代EGFR来解决耐药问题,但是随着新一代药物的使用,新的耐药也会随之出现。PROTAC技术在克服耐药方面已经显现出了一定的优势。Crews、Jian Jin、张三奇、丁克和Gray等课题组都有相关EGFR-PROTACs的研究,旨在通过蛋白降解途径克服耐药突变或找寻突破抑制剂的蛋白降解疗法。可以看出,第一、二、三、四代抑制剂作为结合靶蛋白EGFR的配体,都有被应用到PROTAC的设计中。其中,Gray课题组将变构抑制剂也应用到了PROTAC当中,并取得了不错的效果,能够选择性的降解不同的EGFR突变体,而规避对野生型的降解。体外抗细胞增殖活性也进一步验证了这种选择性。
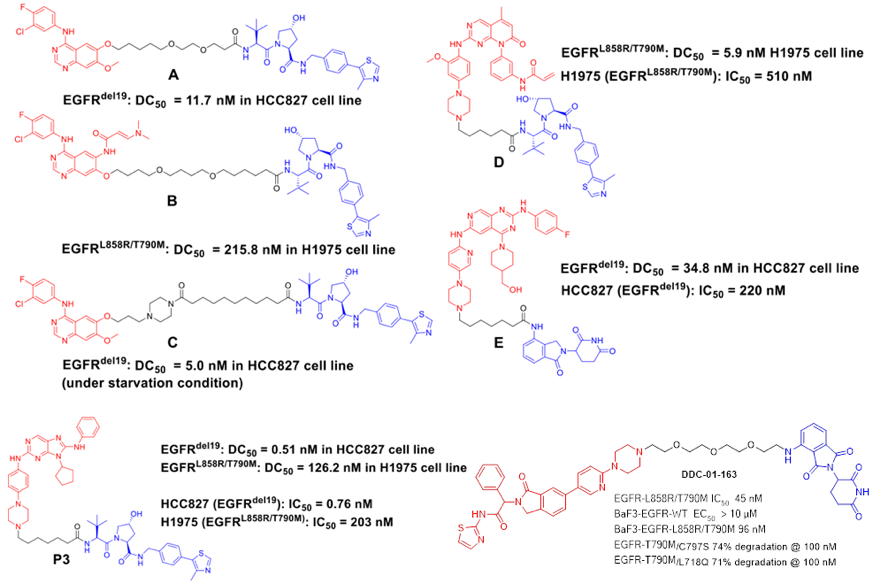
EGFR-PROTACs. [1] Cell Chem Biol 2018, 25, 67-77. [2] J Med Chem 2020, 63, 1216-1232. [3] Eur J Med Chem 2020, 189, 112061. [4] Eur J Med Chem 2020, 208, 112781. [5] Eur J Med Chem 2020, 192, 112199. [6] Angew Chem Int Ed 2020, 59, 14481.
近日,C4 Therapeutics公司对外公布了其EGFR降解剂CFT8919,让在研的第四代抑制剂逊色不少。CFT8919对多种EGFR的突变体都有强效降解活性,包括目前临床无药可用的C797S耐药突变,而且能够规避了对EGFR野生型的降解作用。体内移植瘤模型也验证了该降解剂具有较好的体内抑瘤活性,且对体重影响较小,表明其安全性良好。虽然CFT8919的具体结构未见披露,但是通过分析相关专利可以发现,其所连接的结合EGFR的配体是基于变构抑制剂的,连接的的E3连接酶的配体是基于CRBN的。
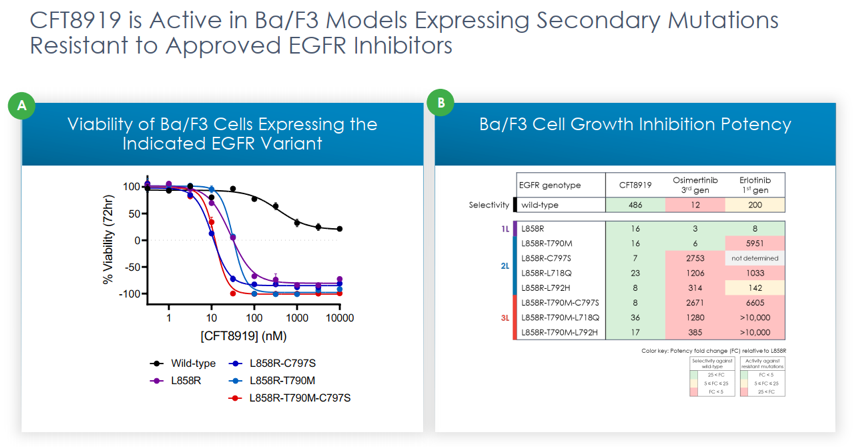
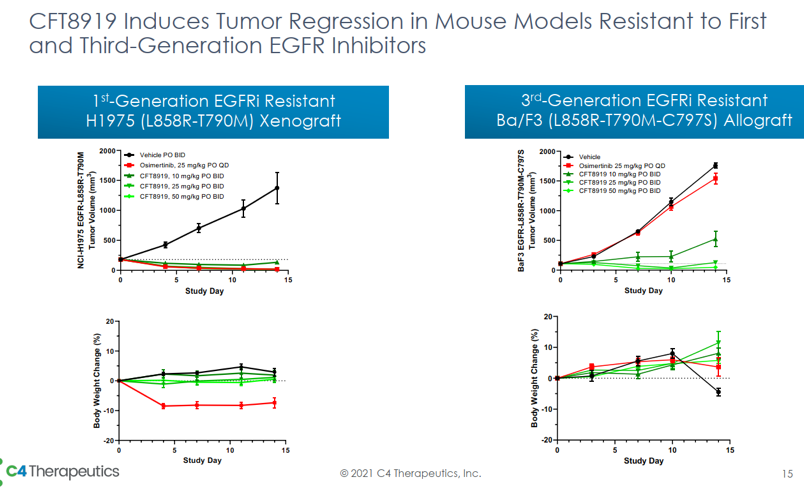
C4 Therapeutics对外公布的数据

WO2021127561A1专利截图
无独有偶,在克服耐药性方面PROTAC在其他靶点上也有所表现。饶燏和Crews课题组针对BTK-C481S耐药突变,基于上市药物Ibrutinib分别得到了强效BTK降解剂P13I和MT-802。不仅可以降解野生型BTK,对BTK-C481S也具有强效降解活性。这为PROTAC技术在克服药物耐药性方面也提供了强有力的支撑。
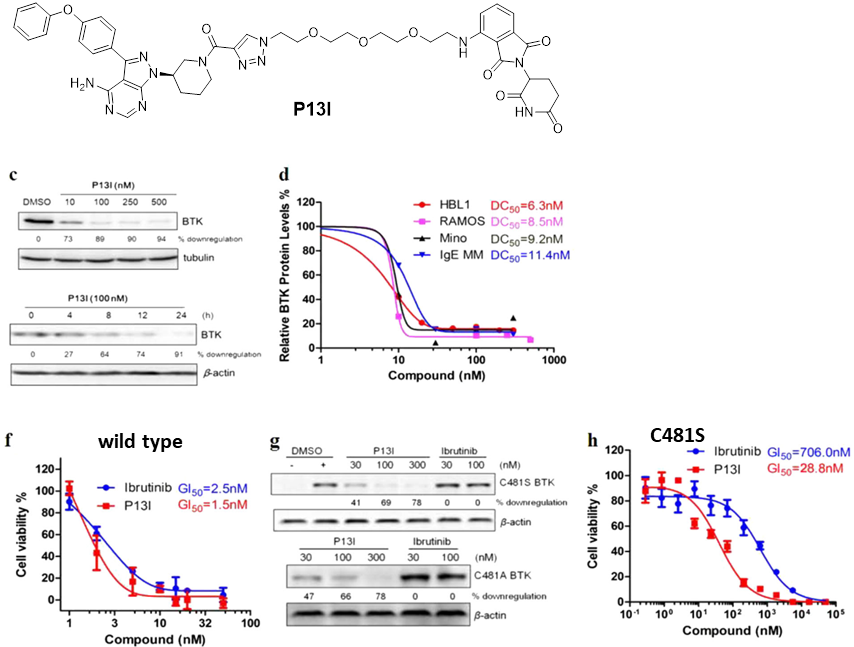
Cell Research 2018, 28, 779-781.
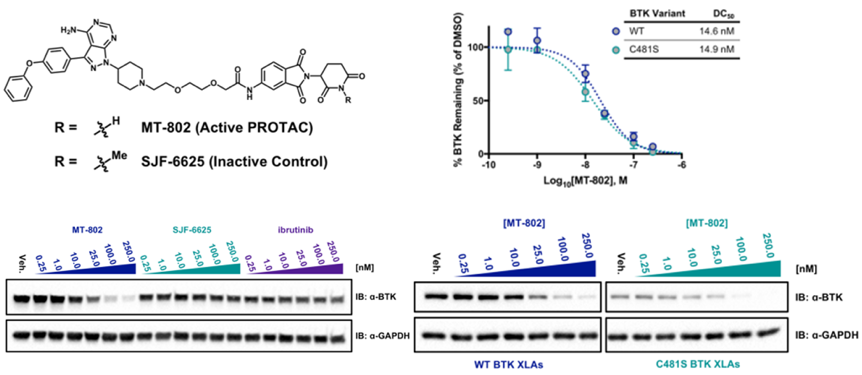
Biochemistry, 2018, 57 (26): 3564-3575.
(4). 简单易用,无需转染
不同于siRNA等基于核酸的蛋白敲除技术,PROTAC是有机小分子,因此无需转染或表达载体就可以轻松地穿透细胞膜,并进入细胞质中发挥作用。因此理论上它可作用于多种细胞系,不要求细胞具有易转染性。
(5). 可以在低剂量下发挥作用
传统的小分子抑制剂需要维持较高的药物浓度,才能持续稳定的占据靶蛋白的活性位点,从而起到抑制作用。而PROTAC分子类似于化学反应的催化剂,降解靶点的同时自身并不会被消耗掉,所以较少的剂量即可发挥作用,这也避免了高剂量药物导致的脱靶毒性。
▉ PROTAC降解的前提—三元复合物的形成
PROTAC介导的蛋白降解是UPS依赖的,而POI-PROTAC-E3三元复合物的形成是靶蛋白被成功降解的前提。目前,“钟”形模型常用来描述三元复合物的形成。文献里经常提到的hook effect也可以用该模型来解释,即PROTAC在较大浓度的时候优先形成的是POI-PROTAC和E3-PROTAC两种二元复合物,影响三元复合物的形成,所以PROTAC的降解活性并不是严格的浓度依赖性,在较大浓度时会出现降解活性下降的反常现象。三元复合物的形成并不是严格的与PROTAC的亲和力呈正相关,因为其也受到PPI(蛋白蛋白相互作用)的影响。协同效应(cooperativity,α)常被用来描述POI和E3之间相互作用对三元复合物稳定性的影响。当α大于1时,POI和E3是正协同效应,对三元复合物的形成有利,反之则不利。
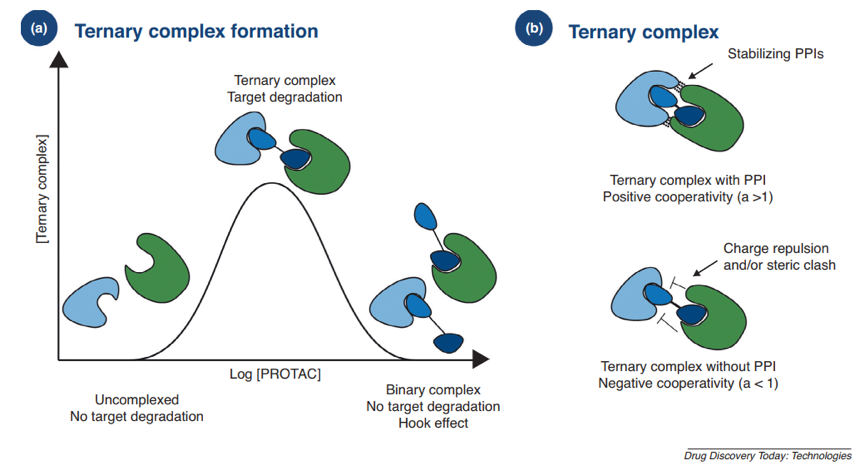
Drug discovery today. Technologies 2019, 31, 15-27.
2017年,Ciulli课题组首次解析了第一个三元配合物BRD4-MZ1-VHL的晶体结构。晶体结构揭示了BRD4与VHL以及PROTAC的Linker与BRD4之间的相互作用。而且通过多种生物物理方法评价了PPI的正协同效应,并证明了这种正协同效应是PROTAC能够诱导BRD家族蛋白选择性降解和高效降解的原因。
 Nat. Chem. Biol. 2017, 13, 514-521.
Nat. Chem. Biol. 2017, 13, 514-521.
▉ PROTAC的设计
PROTAC因为其独特的优势和作用机制,受到广泛关注。如何设计并通过高效的评价体系来发现高效的PROTAC是新药开发的重中之重。尽管目前PROTAC被研究的很多,涉及的靶点也很多,但是构效关系仍然不是很明确。从PROTAC的结构特点出发,其设计的考虑因素主要有以下几点。
(1)POI配体的选择 POI配体一般选择上市或者文献报道的具有一定活性的抑制剂,按照抑制剂能否形成共价键,可分为可逆抑制剂、共价不可逆抑制剂和共价可逆抑制剂。根据结合口袋的不同,又可分为ATP竞争性抑制剂和变构抑制剂。为了获得具有知识产权的PROTAC,常常先对抑制剂做一定的结构衍生,使用优化后的抑制剂作为POI的配体。 (2)E3连接酶及其配体的选择 目前文献报道的应用到PROTAC中的E3连接酶主要有CRBN、VHL、cIAP和MDM2。效果较好、使用频次最高的E3连接酶主要是CRBN和VHL两种。其中,CRBN的配体主要是来那度胺、泊马度胺及其类似物,VHL的配体主要是VHL-L。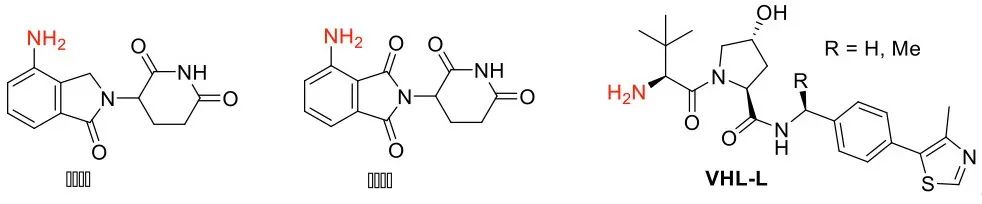
连接酶配体分子
(3)Linker的选择
根据Linker构成的不同,可分为烷基链和PEG链,也有文献使用刚性更强的炔基双哌啶环、含氮原子的螺环或桥环等作为Linker,以限制PROTAC分子的柔性和自由度。已有的文献报道表明,Linker的长短也会影响PROTAC的降解活性,常用的Linker长度一般在4~15个碳原子(或杂原子)。根据作用靶点的不同,Linker的长短对降解活性的影响也不同。此外,Click chemistry(点击化学)由于反应条件较温和、效率较高,常常被应用到PROTAC分子的Linker当中,用于连接两端的分子。
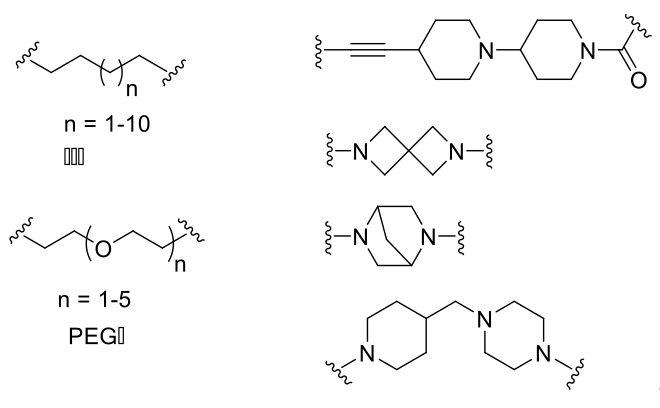
PROTAC中常用的Linker
(4)连接位点的选择和连接点的构成
PROTAC结构上是通过Linker连接两个配体,除了Linker的构成和长短会影响降解活性外,Linker连接的位点也会影响降解活性,甚至影响选择性。POI配体和E3连接酶配体的连接位点一般是在配体暴露在溶剂的区域(图中红色标记,以EGFR为例)。连接位点一般是通过酰胺键、碳原子或杂原子(如O、N等)等连接,通过缩合反应或亲核取代反应等来实现连接。最近,Michael D. Burkart等人发表在J Med Chem的文章,阐述了接头设计在PROTAC中的作用。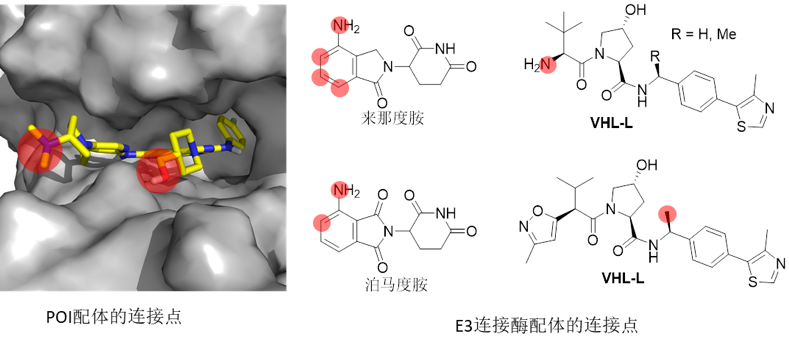
PROTAC连接位点的选择(左图,以EGFR为例说明POI配体连接点的选择;右图,E3连接酶配体连接位点的选择)
▉ PROTAC面临的挑战
虽然PROTAC为新药研发打开了新篇章,给工业界和学术界带来了前所未有的机遇,但同样也面临着诸多挑战。以下我们简要总结了PROTAC发展过程中需要进一步解决的一些问题。
1. PROTAC分子量一般在700~1200道尔顿之间,这使得它们的透膜能力与(口服)生物利用度较差。
2. PROTAC发挥药效必须与目标靶蛋白和E3泛素连接酶结合形成三元复合物。由于HOOK效应(如图所示),PROTAC分子太多或太少都不利于三元复合体的形成。因此,临床上PROTAC分子的使用剂量选择十分困难。
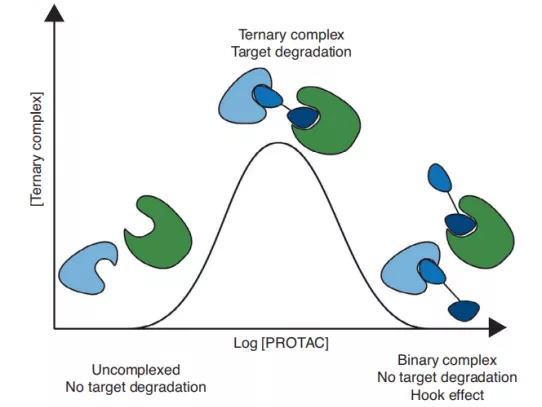
图:hook效应原理
3. 现有的PROTAC技术多基于细胞内的泛素-蛋白酶体降解途径,而泛素-蛋白酶体途径主要作用于胞内蛋白,因此膜蛋白及分泌蛋白的降解仍然是一个巨大的挑战。
4. PROTAC与靶蛋白和E3连接酶的结合仍然以经验为主,如何合理地设计PROTAC分子目前仍缺乏相应的理论基础指导。
5. 目前针对非成药靶点的PROTAC还鲜有报导,未来还需要更多的案例来佐证PROTAC在非成药靶点方面的优势。
6. PROTAC分子评价手段是通过Western Blot的细胞评价,相对周期较长、通量低,从而使PROTAC分子的优化变得困难。
▉ 新一代PROTAC药物开发的机遇
近两年,PROTAC公司备受资本青睐,千万级美元以上融资案例亦不胜枚举,侧面也反应出风险资本对PROTAC赛道的重视与认可。不过在PROTAC之前,有一类蛋白降解药物已经获得FDA批准治疗多种血液癌症,只不过科学家们最近才发现它们作用的机理主要依靠降解与疾病相关的特定蛋白。这就是度胺类药物,包括沙利度胺、来那度胺和泊马度胺。度胺类药物成功的开发经验无疑能够给新一代PROTAC药物的开发带来重要的启示。虽然PROTAC相比于小分子抑制剂,能够更好的克服由于靶标蛋白点突变产生的耐药性问题,但度胺类药物在治疗多发性骨髓瘤等血液瘤患者时,研究人员同样观察到了耐药性问题。理解耐药性的产生无疑对开发下一代更有效的PROTAC药物至关重要。
从度胺类药物的经验来看,肿瘤细胞可以通过多个途径产生对PROTAC药物的耐药性。一是通过降低E3泛素连接酶的活性。已有研究显示,长期暴露在来那度胺之下的多发性骨髓瘤细胞会通过E3泛素连接酶CRBN的突变来降低其酶活性。这些突变最终可能导致三分之一的多发性骨髓瘤患者对来那度胺产生耐药性。另外一种耐药性产生的机制是通过提高E3泛素连接酶的其他底物的水平,通过竞争作用,从而间接保护致病蛋白不被降解。
事实上,人体中有超过600种E3泛素连接酶,而目前大多数的PROTAC分子使用的泛素连接酶仍然局限于CRBN或VHL等少数几种。其中的原因之一是CRBN已经在度胺类药物的实践中得到验证,因此针对这一泛素连接酶设计的靶向蛋白降解剂的安全性较高。然而这也可能更容易造成肿瘤产生耐药性,而且某些致病蛋白并不容易被CRBN或VHL介导的机制降解。如何拓展可用于PROTAC技术的E3泛素连接酶也是PROTAC所面临的挑战之一。
毋庸置疑,PROTAC是小分子药物研发领域内一项革命性的新技术,未来这个领域必将是机遇与挑战并存。我们相信,在学术界和工业界广大同仁的共同努力下,PROTAC技术必将带来药物研发的新时代,解锁新的医学机遇。
▉ PROTAC的先驱、领军人物、相关公司和投资
任何一种新兴技术的发展都离不开相关个人和团体的不懈努力,PROTAC当然也不例外。Crews、Bradner、Ciulli和王少萌等人对PROTAC技术的推进都做出了巨大贡献。PROTAC技术的先驱和领军人物纷纷创立公司,开发蛋白降解技术。国外做的比较早和比较好的公司有Arvinas、C4Therapeutics、Kymera Therapeutics、Vividion、Nurix、Oncopia Therapeutics等。

PROTAC先驱和领军人物
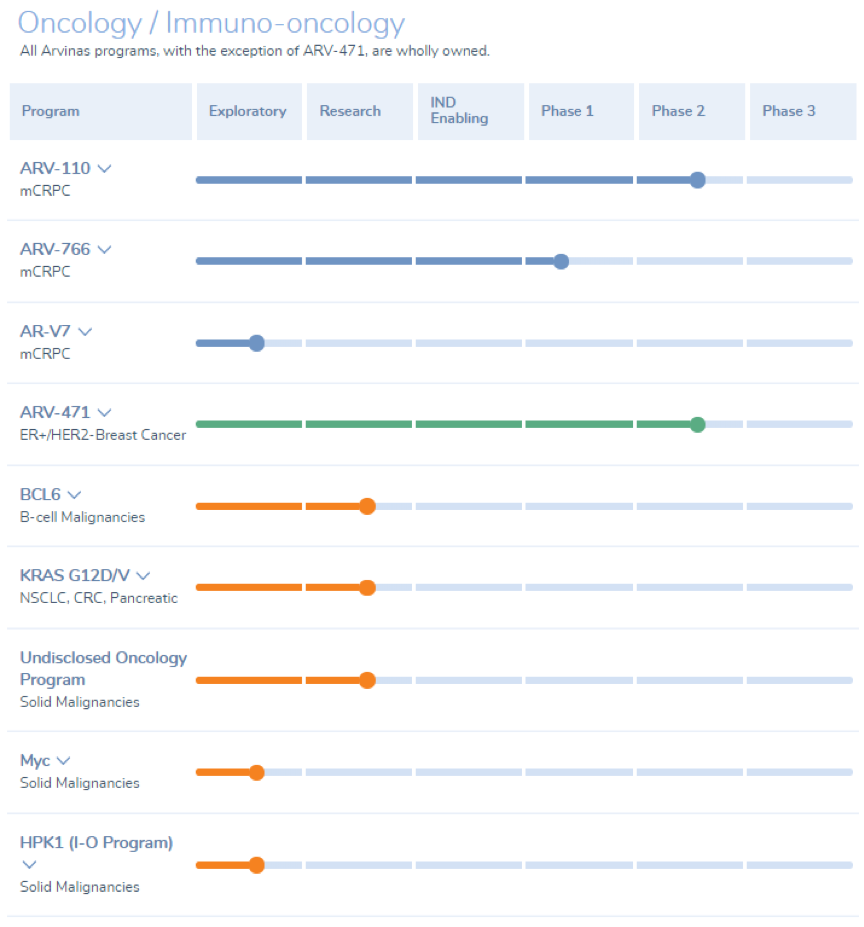
Arvinas公司,由Crews在2013年创立,是最早布局PROTAC的公司之一,开发的蛋白降解技术主要用于肿瘤和神经系统类疾病的治疗。目前进展最快的ARV-110和ARV-471都处在II期临床试验,分别用于前列腺癌和乳腺癌的治疗。神经退行性疾病(如AD、帕金森等)等中枢神经类疾病的治疗效果目前临床非常有限,Arvinas公司针对该类疾病相关的蛋白(如Tau蛋白等)也有布局,期待能够有所突破。

2021年7月22日,辉瑞与Arvinas达成协议,共同开发并商业化ER降解剂ARV-471。根据协议,Arvinas将获得6.5亿美元的前期付款,以及长达14亿美元的里程碑付款,辉瑞还将对Arvinas进行3.5亿美元的股权投资。早在2018年1月辉瑞就与Arvinas达成8.3亿美元的合作协议。除了受到辉瑞的青睐之外,Arvinas还与默沙东、基因泰克、拜耳等制药巨头建立了合作关系。2015年4月,与默沙东达成了4.3亿美元的合作协议。2017年11月,与基因泰克达成了6.5亿美元的合作协议。可以看出,以PROTAC为代表的的蛋白降解疗法正在创造越来越大的价值。

Arvinas官网
C4 Therapeutics,成立于2015年,创始人是James Bradner(目前领导诺华PROTAC的研发)。目前布局的靶点主要是跟肿瘤相关的,比如IKZF1/3、BRD9、EGFR、BRAF-V600E和RET等。该公司拥有专注于蛋白降解剂开发的平台C4T TORPEDO,用于PROTAC的设计、合成和活性评价,旨在发现高质量的蛋白降解剂。2019年1月,与百健和罗氏分别达成4.15亿美元和9亿美元的合作协议。

Kymera Therapeutics,成立于2016年,专注于用蛋白降解技术治疗癌症和免疫性炎症,布局的靶点有IRAK4、STAT3等。进展最快的是KT-474,目前在I期临床。该公司的合作伙伴主要是赛诺菲和葛兰素史克等。2020年7月,与赛诺菲达成多项计划的战略合作,获得1.5亿美元的预付款,并可能获得超过20亿美元的潜在开发、监管和销售里程碑,以及可观的特许权使用费。
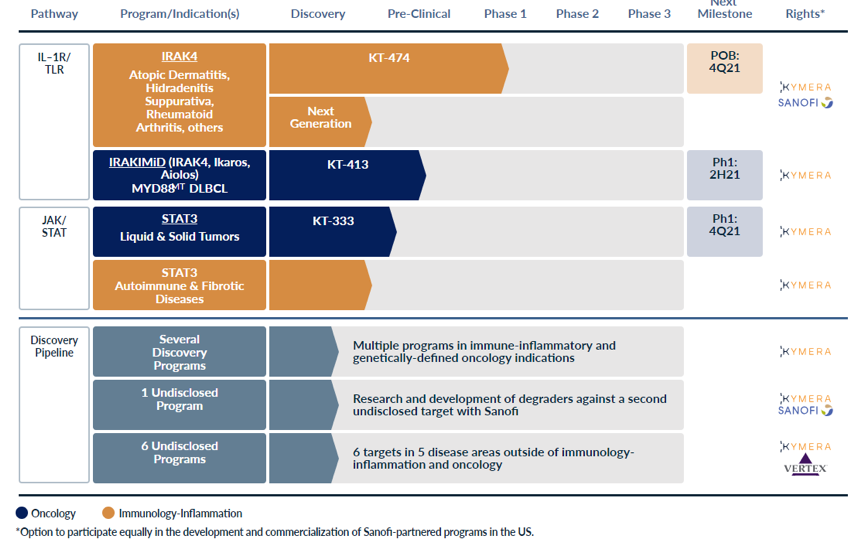 Kymera Therapeutics官网
Kymera Therapeutics官网
Nurix,成立于2009年,专注于开发可供口服的降解剂,布局的靶点有BTK、CBL-B等。该公司利用他们在E3连接酶方面的深厚的专业知识和专有的DNA编码化合物库(DEL),建立了专有的药物发现平台DELigase,用以开发蛋白降解药物或者是抑制E3连接酶来增加有益蛋白的水平。该公司进展最快的蛋白降解药物是NX-2127,目前在临床I期,用于既往治疗失败的B细胞恶性肿瘤。另外一个进展较快的是NX-5948,预计在2021年开启I期临床试验。这两个分子都是口服有效的BTK-PROTAC。Nurix还是目前为数不多的利用PROTAC技术在抗病毒领域有布局的公司之一,虽然靶点未披露,但是这为抗病毒药物的开发带来了新的希望。对于目前造成新冠大流行的SARS-CoV-2病毒,利用蛋白降解疗法或许会有所突破,期待相关研究的报道。Nurix的合作伙伴有赛诺菲和吉利德等。2020年1月,Nurix宣布与赛诺菲进行全球战略合作,针对多种具有挑战性的疾病,开发创新靶向蛋白质降解药物。根据协议,Nurix获得5500万美元的预付款,并在扩大合作靶标的数量后获得后续付款。此外,在成功完成临床前、临床、申报和销售里程碑后,Nurix将有资格获得最高约25亿美元的总付款。2019年6月,Nurix与吉利德达成一项战略协议,利用Nurix的DELigase平台开发能够分解致病蛋白的新型抗癌药物。根据协议,Nurix获得4500万美元的预付款。此外,Nurix与吉利德合作过程中,将机会获得高达23亿美元的里程碑付款以及未来的销售分成。
 Nurix官网
Nurix官网
海外布局PROTAC赛道的公司除了初创的Biotech外,诺华、BMS、安进等制药巨头也纷纷加入其中。
PROTAC技术因其颠覆性的理念,国内本土公司也有布局,并有可能赶超海外药企的领域。据不完全统计,国内已布局PROTAC技术的药企已超过20家,不仅有biotech,还有big pharma。海思科作为第一家国内开启临床试验的公司,位列PROTAC技术第一梯队。其他药企还有凌科药业、分迪科技、美志医药、江苏恒瑞、开拓药业、成都先导、海创药业、海和药物、领泰生物、和径医药、标新生物、诺诚健华、五元生物、美志医药、亚盛医药、科伦药业、石药集团、嘉兴优博、上海睿因、苏州德亘生物、分迪科技、多域生物、鲁南制药等。具有PROTAC开发平台的CRO公司有药明康德、康龙化成、美迪西等。
海思科,具有比较完善的PROTAC平台,用以开发针对肿瘤和自身免疫性疾病的高选择性且口服有效的蛋白降解药物。目前PROTAC的项目有超过20个,布局的靶点有BTK、IRAK4、BRD4、AR、EGFR、KRAS、ALK、CDK4/6、EZH2、PARP、Bcr/Abl、STAT3等。其中以BTK-PROTAC(研发代号HSK29116)进展最快,2021年1月获批临床,4月启动临床,是国内首个进入临床的PROTAC药物。与已上市BTK抑制剂相比有可能解决BTK-C481S耐药,同时具有潜在的更优的药效、更好的选择性和更小的副作用。
开拓药业,2021年2月,宣布其自主研发的全球首个基于PROTAC技术的外用AR降解剂(GT20029)的IND申请已获国家药品监督管理局受理。据了解,开拓药业早在2018年下半年就正式启动基于PROTAC的AR降解剂的研发。该公司将通过开发非口服的PROTAC药物来验证这一开创性的技术是否可以成药,同时也正在研发口服的靶向AR的PROTAC。其选择的适应证为雄激素性脱发和痤疮等。
江苏恒瑞,作为国内创新药龙头,在PROTAC领域也早有布局。早在2019年,恒瑞和上海宏创医疗科技有限公司就联合收购了Princeps Therapeutics 31%的股权,进军PROTAC领域。目前PROTAC已经成为恒瑞的重要研发方向。近日,恒瑞的ER-PROTAC的专利被公开,专利中活性最好的化合物是12号化合物,对ER的DC50和对MCF-7的抗增殖活性都在亚纳摩尔级别,对携带ER-Y537S突变体的MCF-7也具有强效抗增殖活性,IC50为1.27 nM。该化合物为ARV-471的Me-too,在CRBN配体来那度胺的基础上采取了并环的策略来突破已有的专利。

恒瑞公开的ER-PROTAC专利代表化合物12,专利WO2021143822A1
亚盛医药,2020年11月,亚盛医药与密歇根大学达成协议,获得MDM2降解剂的全球独家权益。目前,该降解剂已经进入IND申报阶段。根据已有文献报道,推测该次达成协议的MDM2的降解剂可能为MD-224,该分子来自王少萌课题组。王少萌是PROTAC技术的领军人物,同时也是亚盛医药共同创始人兼首席科学家顾问。
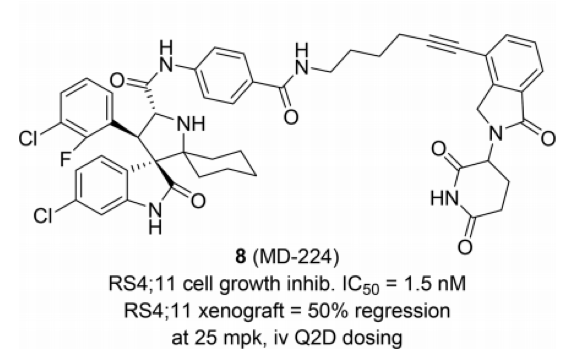
亚盛医药与王少萌合作的MDM2降解剂可能的结构(J Med Chemistry 2019, 62, 448-466. J Med Chemistry 2019, 62, 445-447.)
百济神州,作为国内研发投入最大的公司,该公司在PROTAC领域研发进展也很迅速。据悉,该公司已经具备PROTAC药物自主研发的平台。2021年8月16日,百济神州在ClinicalTrials.gov上登记了一项BTK降解剂BGB-16673的临床试验,适应症为B细胞恶性肿瘤、边缘区淋巴瘤滤泡性淋巴瘤、非霍奇金淋巴瘤、华氏巨球蛋白血症。这是百济神州首个进临床的PROTAC项目,也是国内第二个进入临床试验的BTK-PROTAC。从已经公开的专利来看,其PROTAC分子应该是基于该公司自主研发的BTK抑制剂泽布替尼(Zanubrutinib)的,E3连接酶的配体是来那度胺及其类似物。
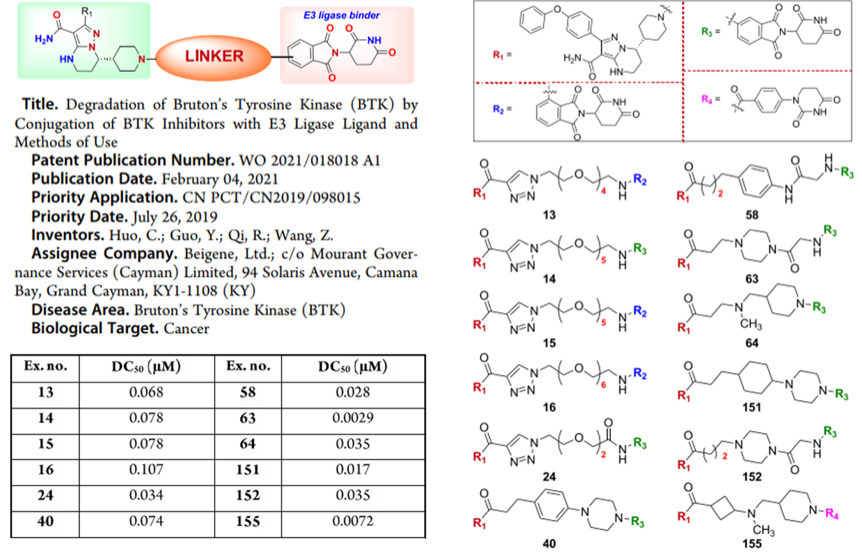
ACS Med. Chem. Lett. 2021, 12, 688−689. WO2021018018A1
▉ 总结
作为革命性技术,PROTAC的发展经历了20年,尤其在过去的5年发展迅猛,俨然已经成为新药研发的新风口。利用该技术可布局的靶点广阔,市场巨大,未来可期。但是PRORAC分子量大、生物利用度差、成药困难的缺点也很明显,期待该技术的不断进步和完善。相信随着成药性差的难题被攻克,PROTAC可以成为像小分子抑制剂、单抗和免疫治疗等一样成功的疗法。当然,我们也期待PROTAC能够上市,用于治病救人,延长患者生命并提高患者的生活质量。

