可防可治的罕见病:21-羟化酶缺乏症
时间:2023-04-22 11:19:12 热度:37.1℃ 作者:网络
什么是21-OHD?
先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是一组由于肾上腺皮质激素合成酶基因突变导致酶活性缺乏或减低,肾上腺皮质激素合成减少,对下丘脑-垂体-肾上腺轴(HPA轴)负反馈作用减弱,促皮质素(ACTH)分泌增多,继发肾上腺皮质增生引起的一组疾病。
21-羟化酶缺乏症(21-hydroxylase deficiency,21-OHD)是CAH中最常见的类型,是由于编码21-羟化酶的CYP21A2基因缺陷导致肾上腺皮质类固醇激素合成障碍的一种先天性疾病,呈常染色体隐性遗传,已被列入中国罕见病目录。
21-OHD有哪些临床分型和临床表现呢?
依据21-羟化酶残留酶活性差异,21-OHD临床分为3种类型:失盐型、单纯男性化型及非经典型。
1. 失盐型(salt wasting,SW,约占75%):最严重的21-OHD表型,无残留酶活性,醛固酮、皮质醇完全缺乏,雄激素生成增多。出生后不久即可出现呕吐、拒食、脱水、代谢性酸中毒、低钠血症和高钾血症等肾上腺危象表现,女性可有外生殖器男性化,若不及时诊治,病死率高。
2. 单纯男性化型(simple virilizing,SV,约占25%):残留酶活性1% -2%,临床无失盐型表现,女性患者出生时即有外生殖器男性化表现,男性患者可出现假性性早熟。
3. 非经典型(non classical CAH,NCCAH):亦称迟发型,21-羟化酶残留酶活性20% -50%。临床表现不特异,可在儿童期或青春期甚至成人期发病,出现阴毛早现、性早熟、月经紊乱、不孕等。
如何筛查21-OHD?
应用放射免疫法检测血清17-OHP水平是21-OHD的一级筛查方法。健康儿童出生后17-OHP可轻度升高,出生1 d迅速下降,因此建议在出生48 h后进行。早产儿17-OHP较足月儿童升高,建议在出生后2 周及4 周进行17-OHP复测。
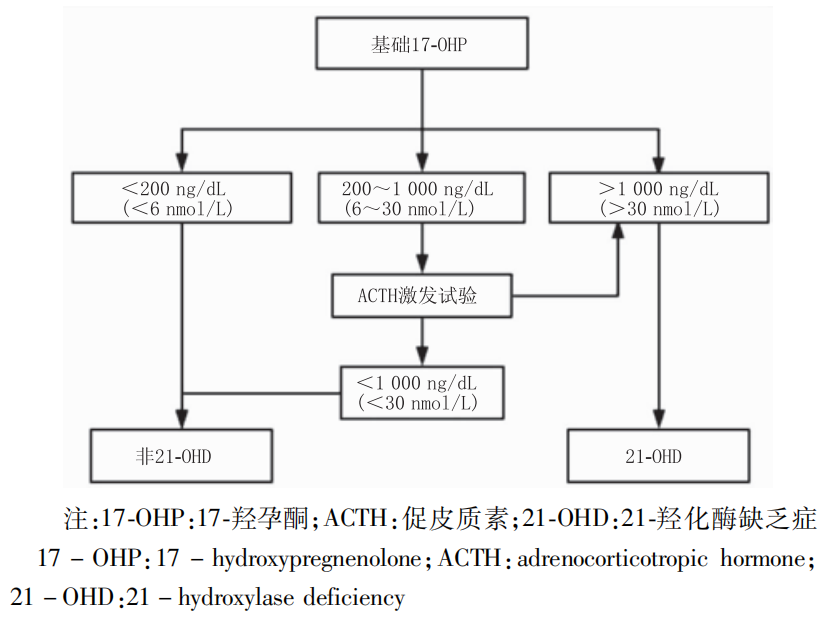
图1 21-OHD诊断流程图(参考自中华实用儿科临床杂志)
筛查阳性患儿或在儿童期有症状怀疑21-OHD患者,需行ACTH 1-24激发试验。于清晨8:00 -9 :00进行,静脉推注 0.25mg ACTH,极低出生体质量儿可减少为0.125 mg,检测基线值及60 min时血清17-OHP水平,如有条件,建议同时进行脱氢表雄酮、雄烯二酮、11-脱氧皮质醇、11-脱氧皮质酮水平测定,相关激素比值可用于鉴别 CAH 类型。
诊断了21-OHD,如何治疗呢?
21-OHD治疗的目的是纠正糖皮质激素、盐皮质激素缺乏及抑制 ACTH 和肾上腺来源的高雄激素过多分泌。治疗包括药物及手术治疗。
1、糖皮质激素替代治疗 21-OHD患者糖皮质激素替代治疗的目的是纠正皮质醇缺乏,但为抑制ACTH及雄激素过多分泌,替代剂量通常高于生理需要量。
2、盐皮质激素替代治疗 尽管仅有失盐型患者具有相关失盐表现,但实际上所有21-OHD 患者均存在盐皮质激素缺乏。对于21-OHD 患者,建议在儿童期加用盐皮质激素进行替代治疗。
3、手术治疗 早期手术有助于恢复正常解剖结构、减少泌尿系统感染发生、降低患者及其父母的心理压力及缓解焦虑情绪,减少心理损伤。
4、其他治疗 包括雄激素水平过高者,可考虑同时加用雄激素受体括抗剂以减轻高雄激素血症;合并中枢性性早熟可考虑应用促性腺激素释放激素类似物治疗,以抑制中枢性腺轴改善终身高;亦包括糖皮质激素泵、糖皮质激素缓释剂的应用。
参考文献:
[1] Speiser Phyllis W,Arlt Wiebke,Auchus Richard J et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline.[J] .J Clin Endocrinol Metab, 2018, 103: 4043-4088.
[2] 陈晓波,高亢. 21-羟化酶缺乏症诊治新进展. 中华实用儿科临床杂志,2019,34(20):1526-1530.
[3] 中华医学会儿科学分会内分泌遗传代谢病学组. 先天性肾上腺皮质增生症21-羟化酶缺陷诊治共识. 中华儿科杂志,2016,54(8):569-576.

