Neurology:线粒体疾病中MT-ATP6/8缺陷的自然史
时间:2025-03-24 12:10:38 热度:37.1℃ 作者:网络
线粒体DNA(mtDNA)基因MT-ATP6和MT-ATP8编码三磷酸腺苷(ATP)合成酶复合物的α亚基和8亚基(A6L)。这些基因的致病性变异会导致一系列无法治愈的线粒体综合征,临床表现多样,包括共济失调、运动和语言发育迟缓、耳聋、视网膜色素变性以及脑部MRI显示的Leigh综合征样病变。尽管较高的mtDNA变异水平通常与更严重的症状相关,但即使携带相似变异负荷的个体,其临床表现也存在显著差异。这使得潜在治疗方法的开发面临巨大挑战。
最近,一项国际多中心研究通过对MT-ATP6/8缺陷患者的回顾性自然史分析,为未来的临床试验提供了关键终点数据。这项研究不仅揭示了疾病的临床特征和生化标志物,还为疾病管理提供了重要依据。

研究团队从意大利、德国、美国和西班牙的国家参考中心收集了111名经基因确诊的MT-ATP6/8缺陷患者的临床、生化和分子遗传学数据。这些数据通过伦理委员会批准的线粒体疾病国家登记系统或本地项目获取。研究分析了患者的发病年龄、临床表现、影像学特征、生化指标以及生存率。
研究结果显示,44%的患者在婴儿期(<1岁)发病,36%在儿童期(≥1岁且≤12岁)发病,20%在成年期(>12岁)发病。Kaplan-Meier分析显示,婴儿期和儿童期发病患者的生存率显著低于成年期发病患者(p = 0.0349),但总体死亡率较低(8%)。在受累组织方面,中枢神经系统(CNS)是最常受累的组织(93%),其次是肌肉(75%)、眼睛(46%)和心脏(18%)。脑部MRI显示,58%的患者存在孤立的Leigh样病变,15%的患者同时存在Leigh样病变和皮质/小脑萎缩,10%的患者仅有小脑萎缩,21%的患者表现为其他病变。在最后一次随访中,11%的患者需要依赖轮椅。
生化标志物方面发现,56%的患者瓜氨酸水平降低,49%的患者丙氨酸水平升高,71%的患者乳酸水平升高,提示这些指标可能作为潜在的生物标志物。分子遗传学发现共鉴定出26种致病性变异,其中6种为新发现。
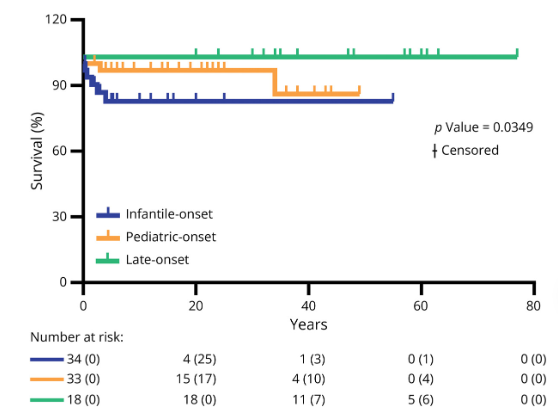
综上,这项研究为MT-ATP6/8缺陷的临床管理提供了重要数据,并提出了基于发病年龄的更精确分类方法。MT-ATP6/8缺陷是一种复杂的线粒体疾病,其临床表现多样且治疗困难。这项国际多中心研究通过回顾性自然史分析,为理解疾病的临床特征、生化标志物和遗传基础提供了重要数据。未来,随着研究的深入和技术的进步,我们有望为MT-ATP6/8缺陷患者提供更精准的诊断和治疗方案,帮助他们改善生活质量。
原始出处:
Garone C. Natural History of Patients With Mitochondrial ATPase Deficiency Due to Pathogenic Variants of MT-ATP6 and MT-ATP8. Neurology. 2025 Apr;104(7):e213462. doi: 10.1212/WNL.0000000000213462.

