【衡道丨病例】Kikuchi–Fujimoto病(组织细胞坏死性淋巴结炎)如何诊断?
时间:2024-10-17 19:00:35 热度:37.1℃ 作者:网络
患者病史
患者女性,20岁,发现颈部淋巴结肿大一月,伴盗汗、淋巴结触痛。
影像检查
B超:
双侧颈部低回声,右侧之一大小:15.1×7.2mm,左侧之一大小:11.0×6.1mm。双侧锁骨上见低回声数个,右侧之一大小:10.8×6.4mm,左侧之一大小:5.5×3.9mm。超声引导下右侧颈部淋巴结行粗针穿刺。
镜下形态
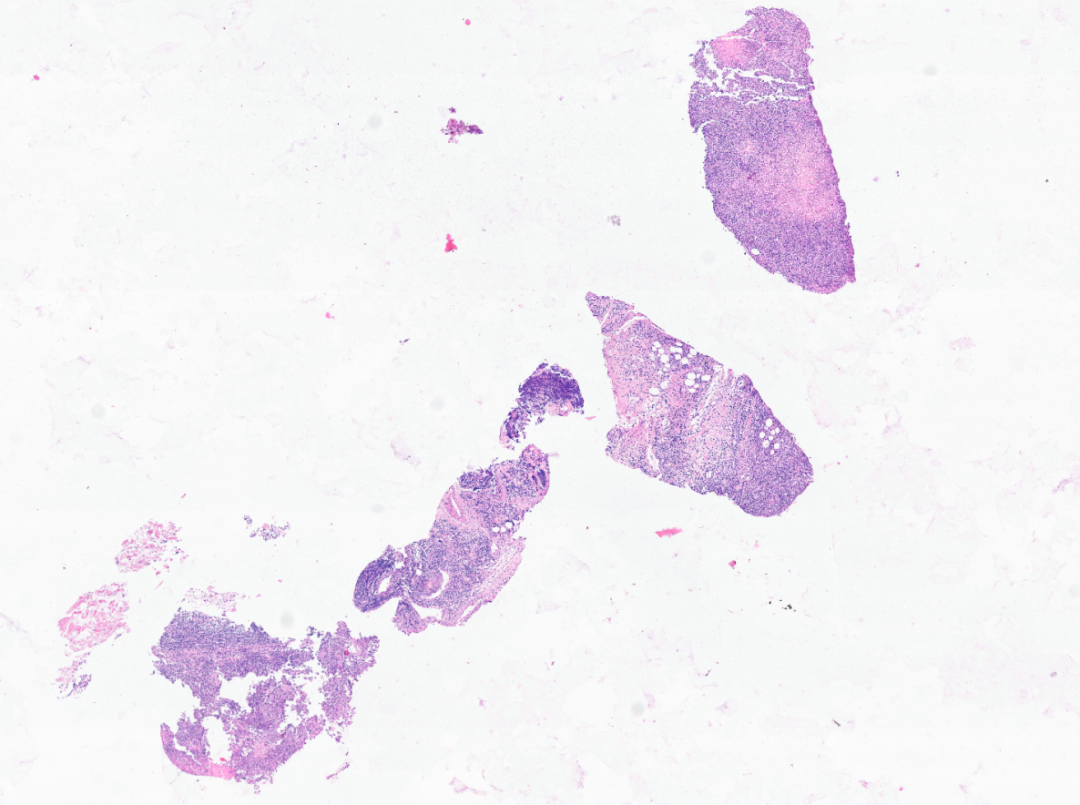
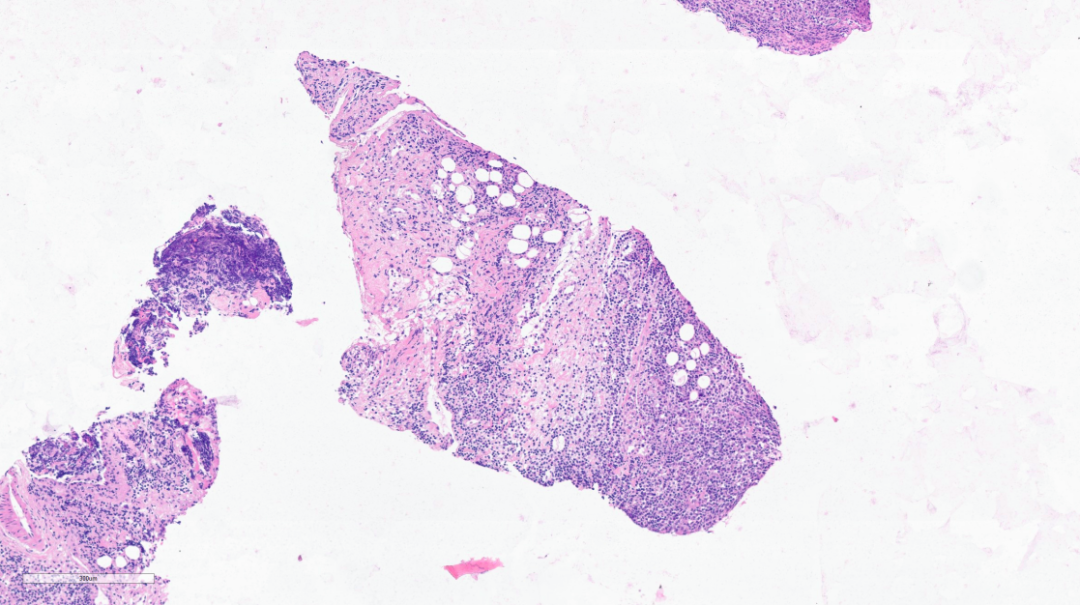
穿刺组织较小,低倍镜下见淋巴结结构被破坏,部分区域淡染
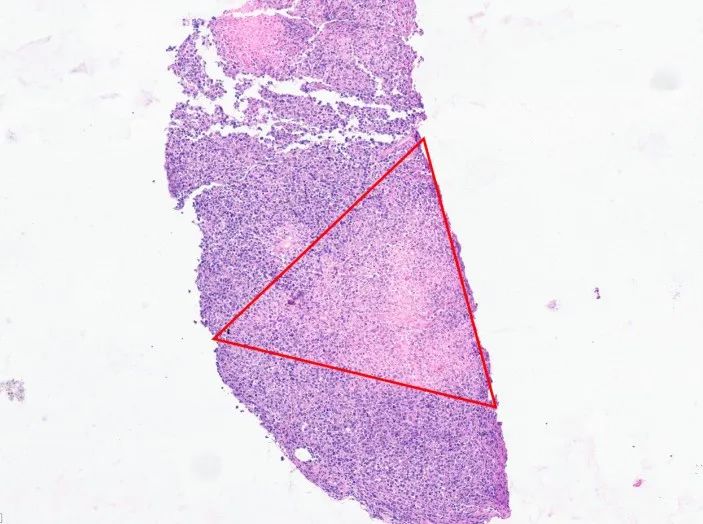
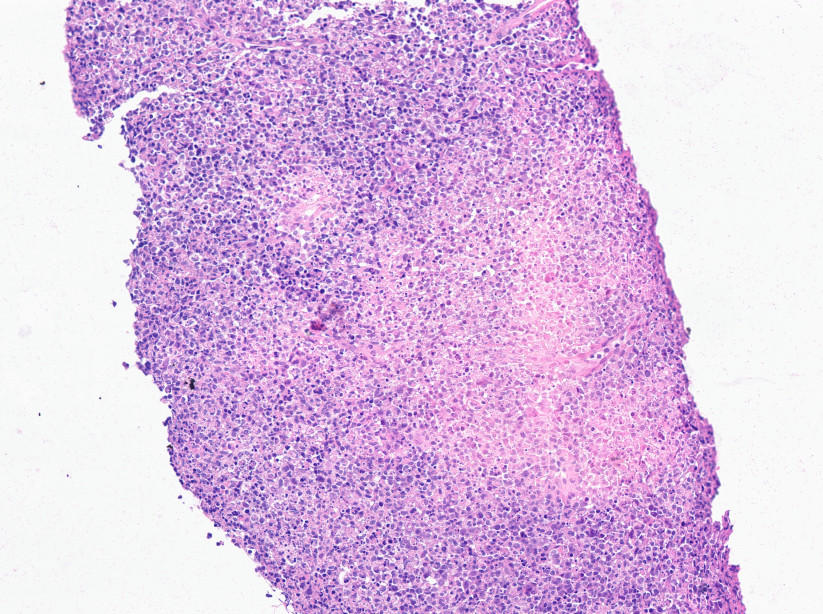
淡染区部分呈三角形,为细胞坏死区域
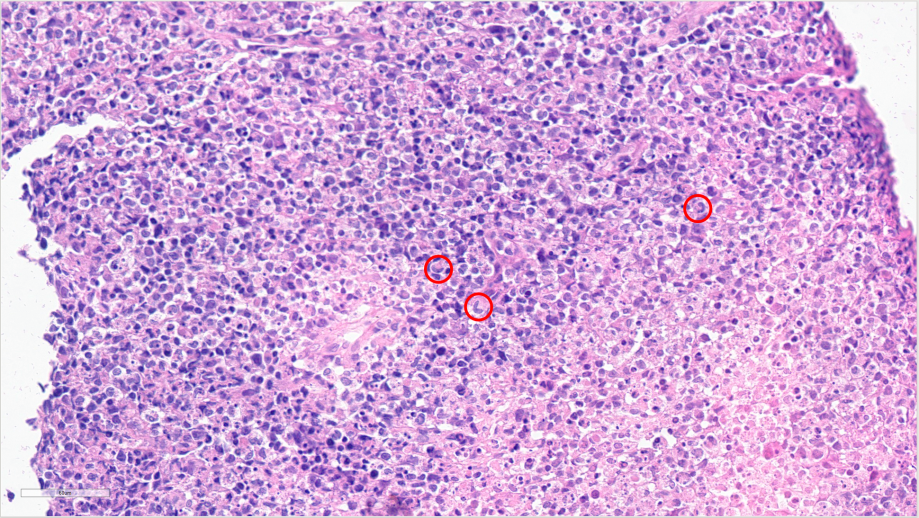
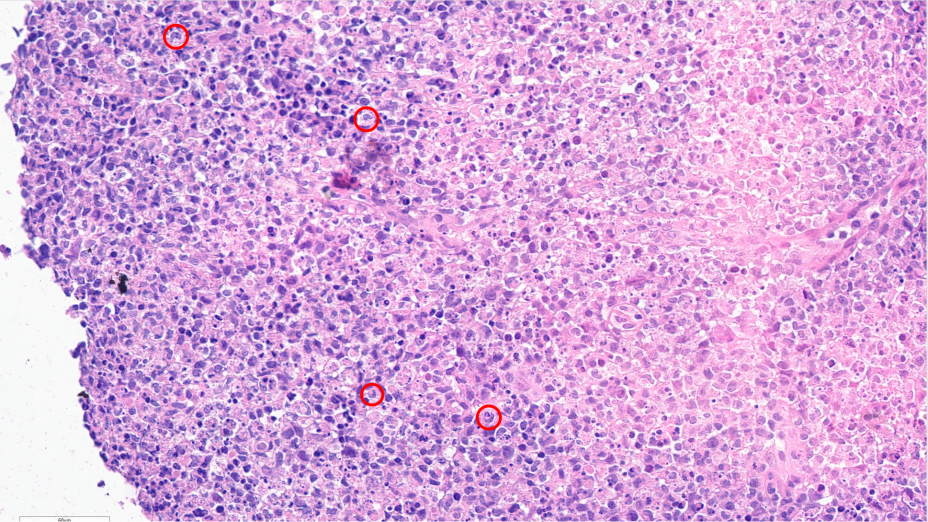
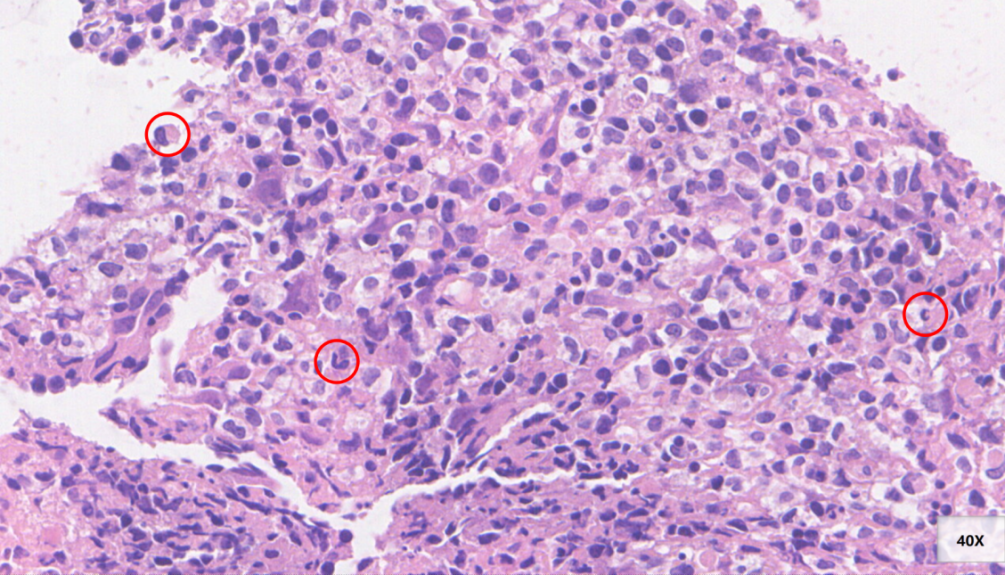
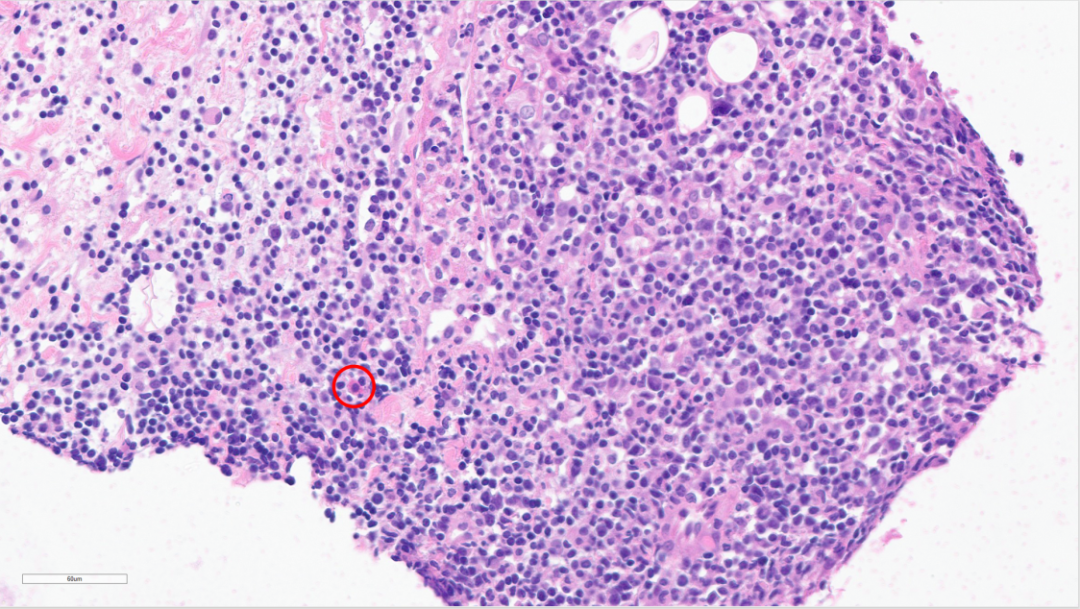
坏死区可见活跃的细胞崩解现象,周边细胞较大,细胞核淡染,可见部分呈肾形、新月形细胞核(红色圆圈)
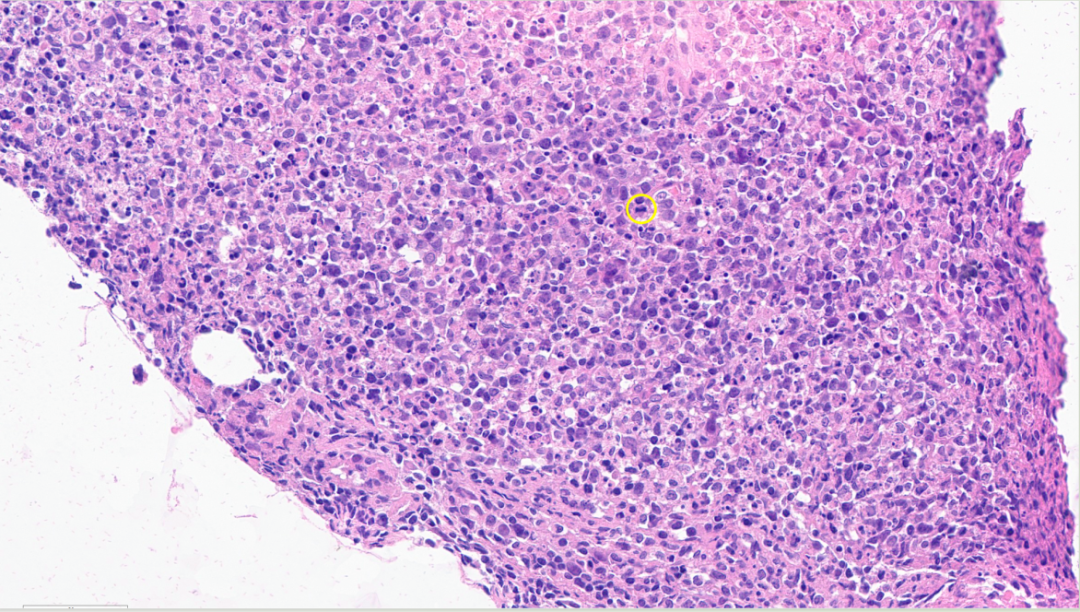
可见核分裂象(黄色圆圈)以及活跃的细胞崩解现象
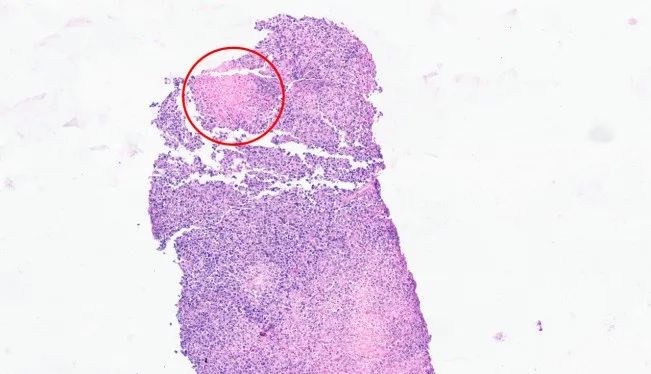
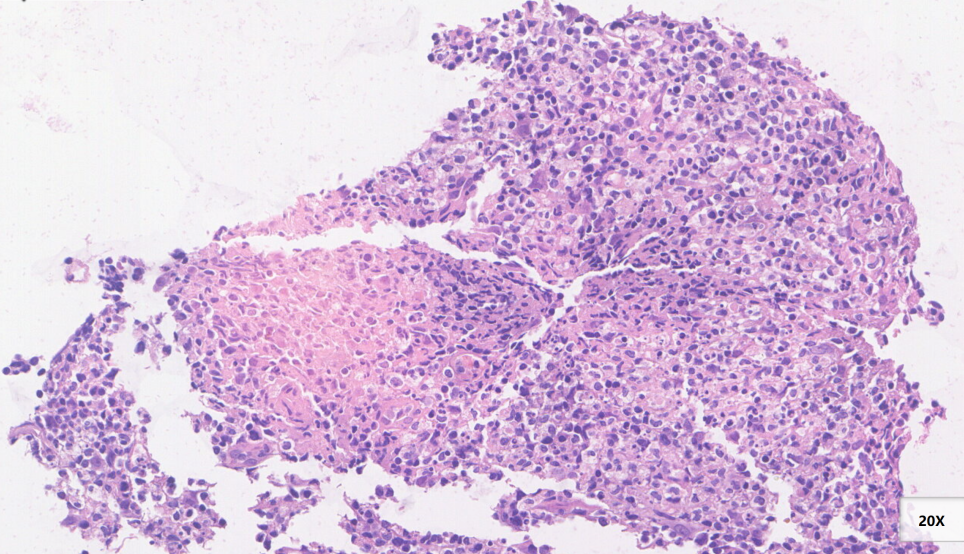
另一片坏死区域(红色圆圈),坏死区周边可见核形不规则细胞
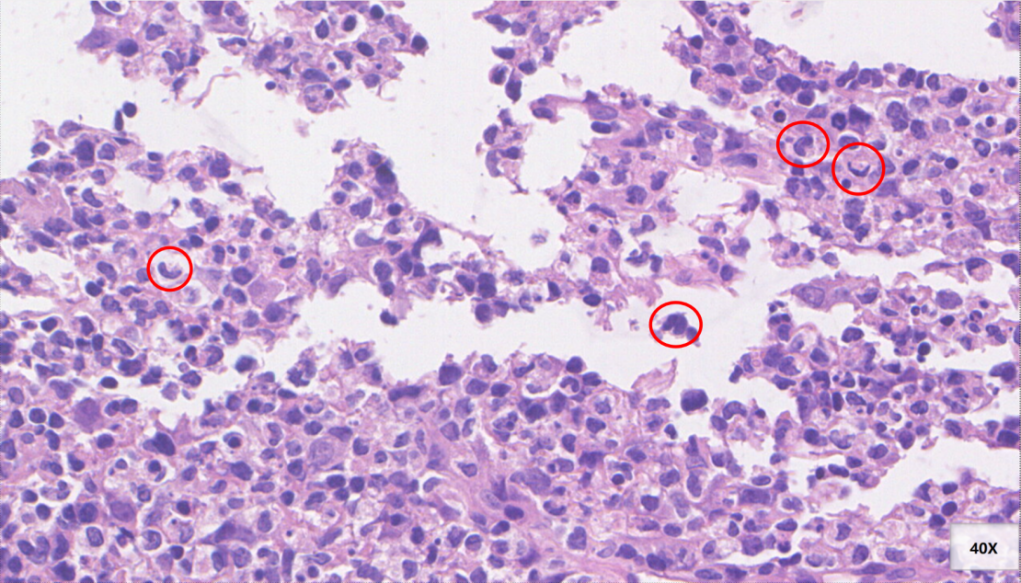
细胞浆丰富,细胞核偏位,细胞核形态不规则,呈新月形、印戒样(红色圆圈)
另一块组织,同样可见核型不规则细胞,另查见极个别嗜酸性粒细胞(红色圆圈)
免疫组化
1、阳性标记:
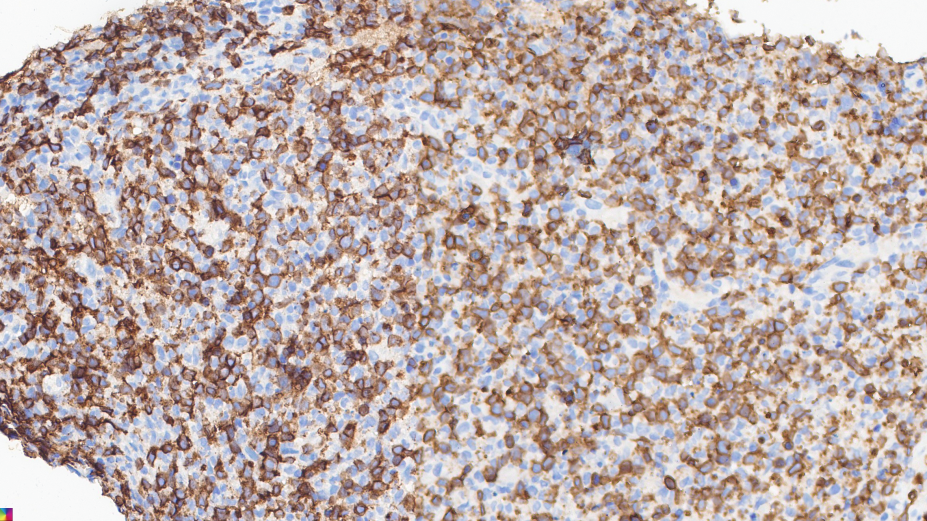
病灶区淋巴细胞 CD3+
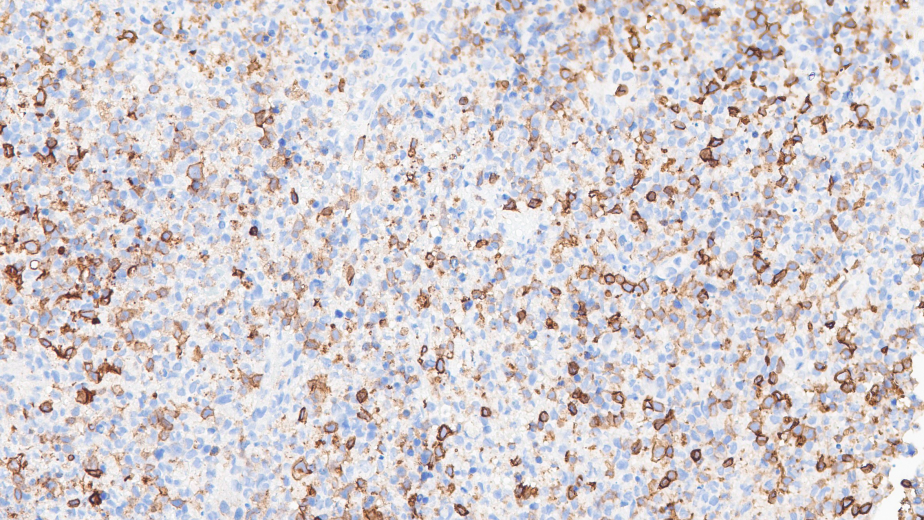
CD5少量+
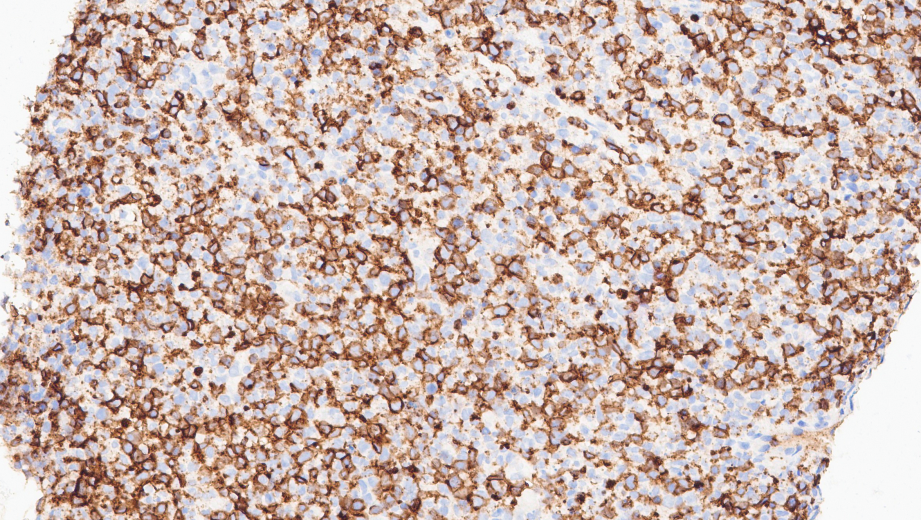
CD2+
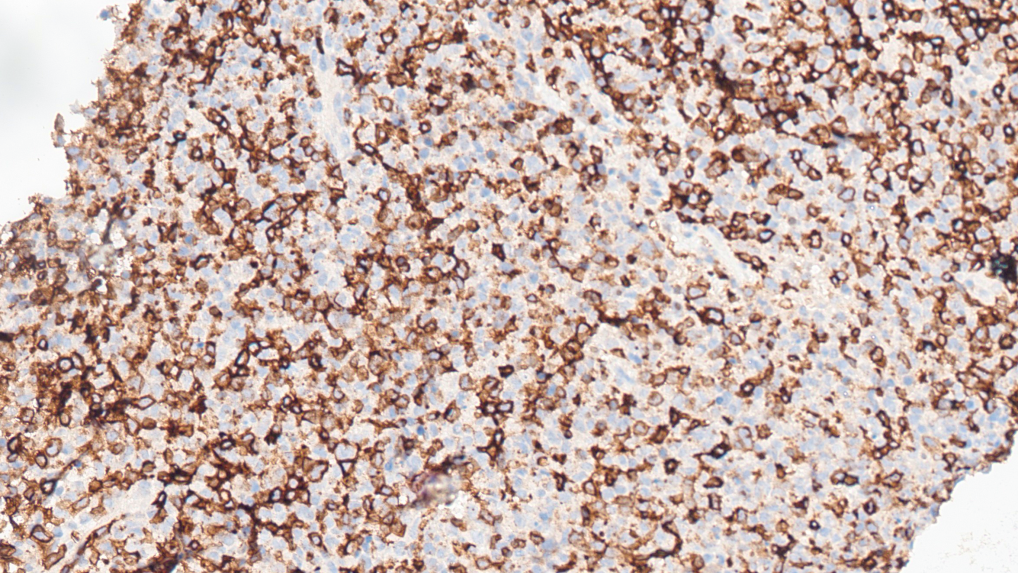
CD7+
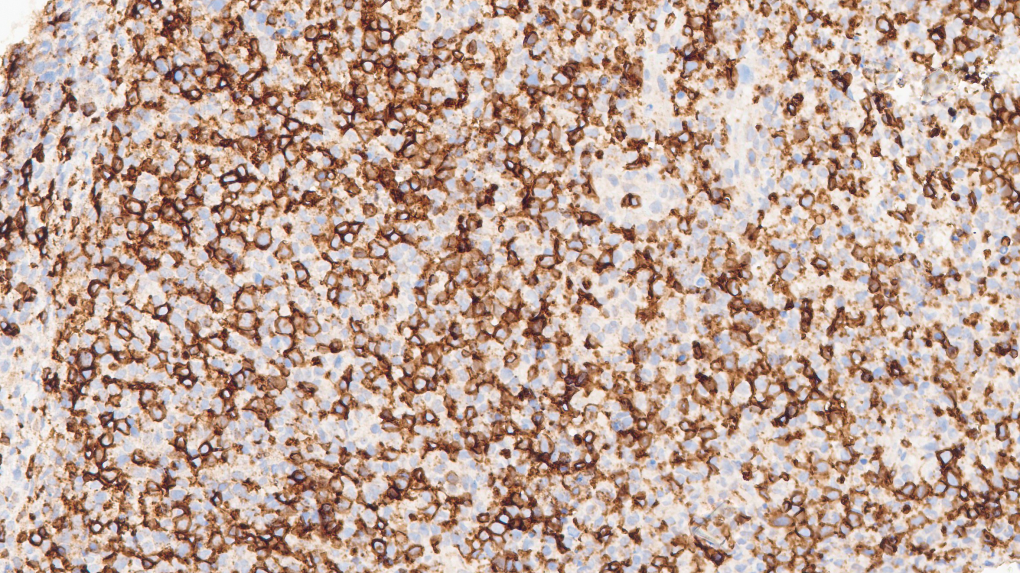
CD8+
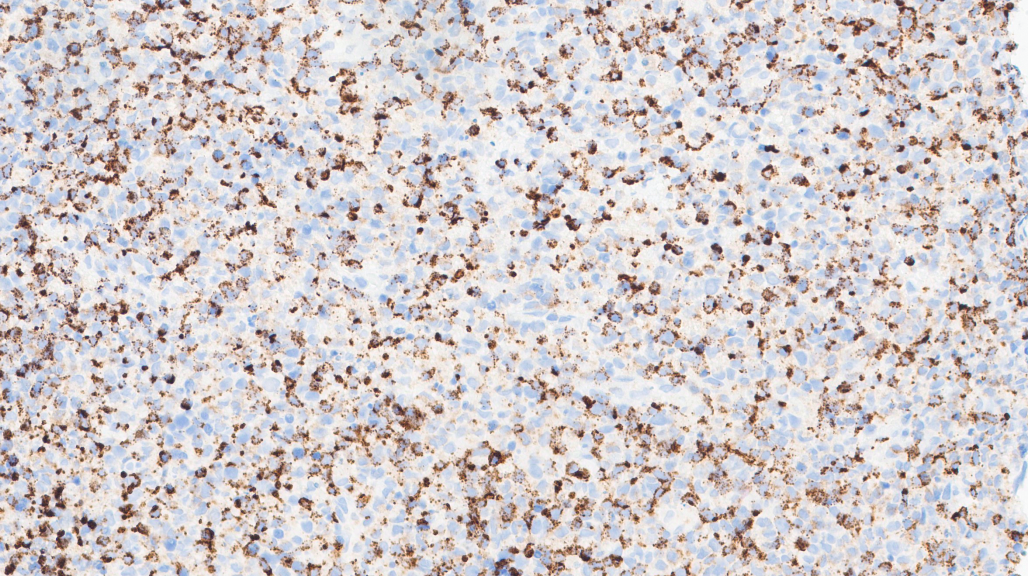
TIA-1+
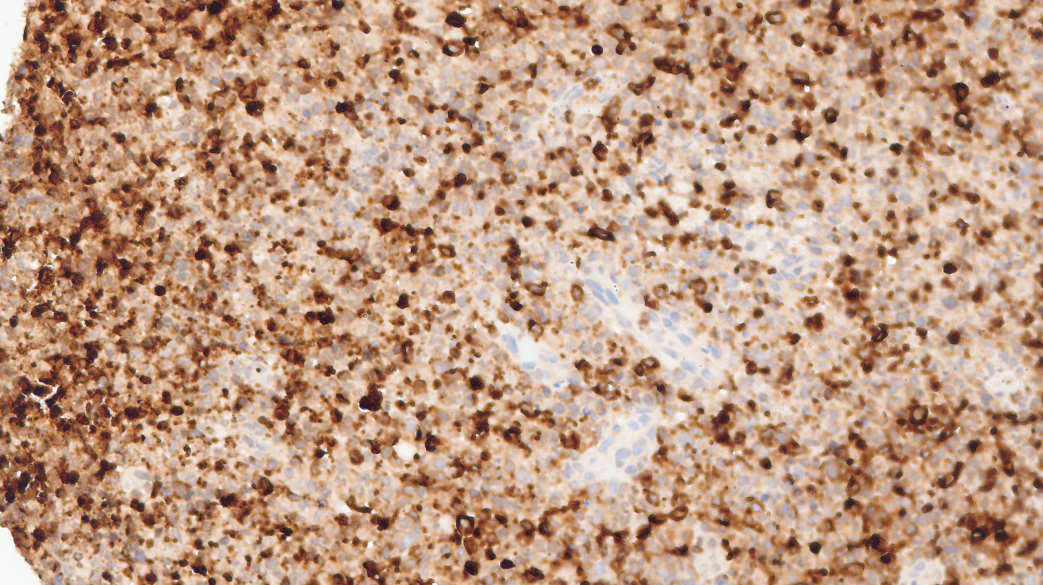
Granzyme-B+
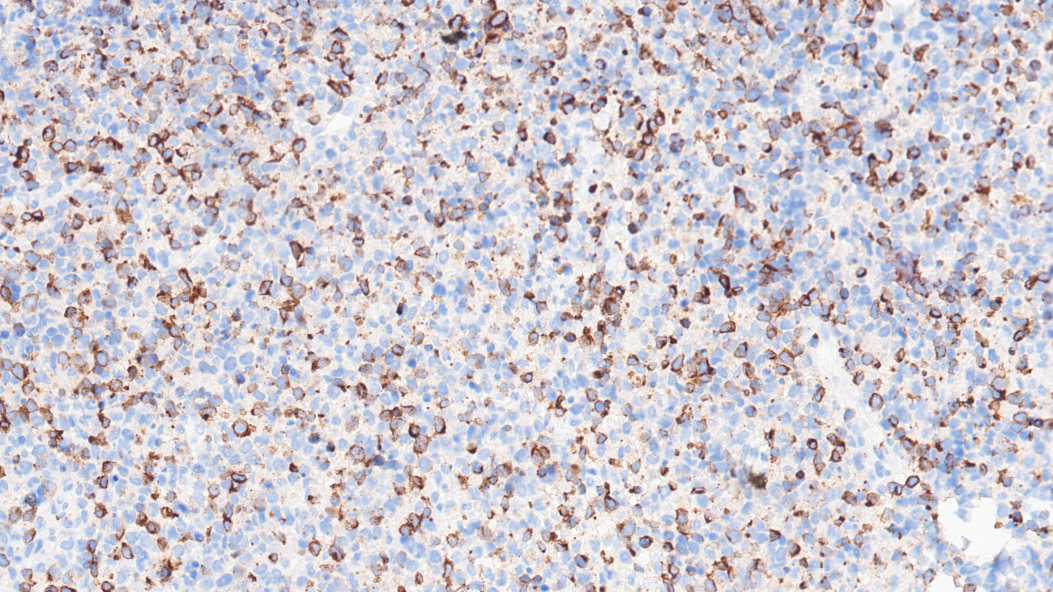
Perforin+
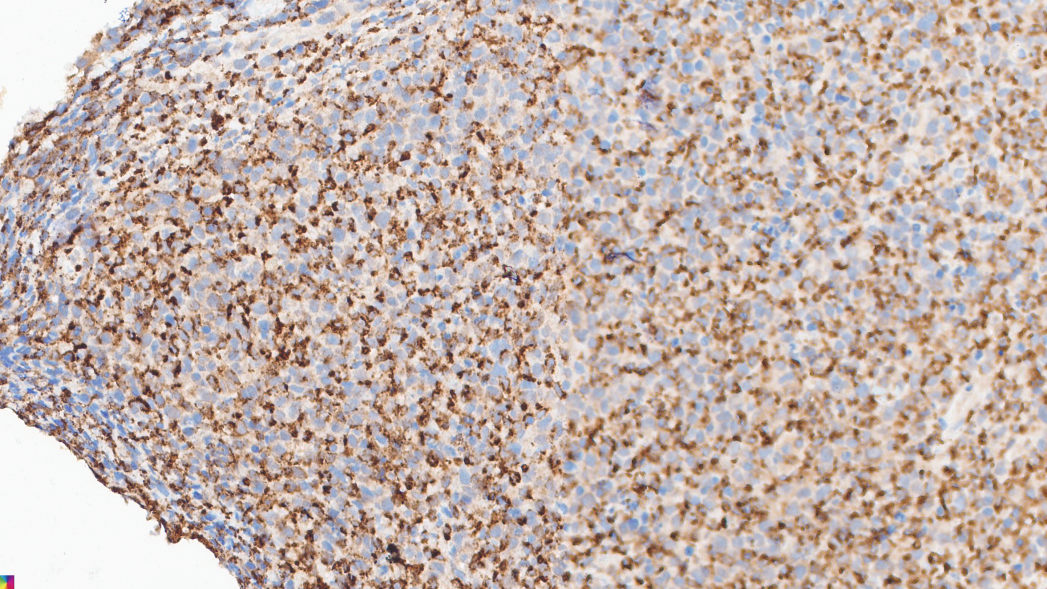
组织细胞:CD68+
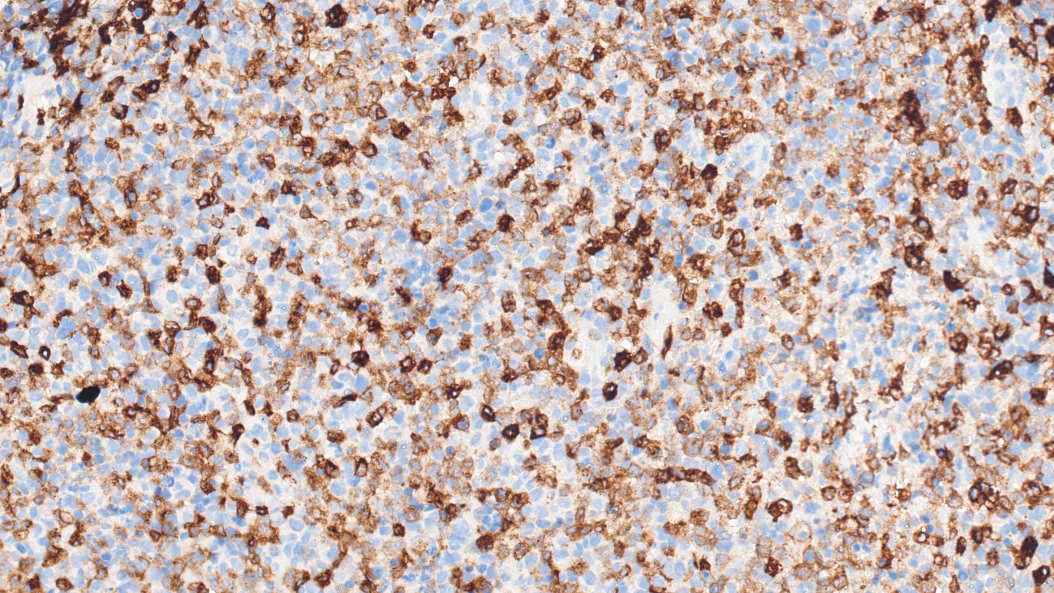
组织细胞:CD163+
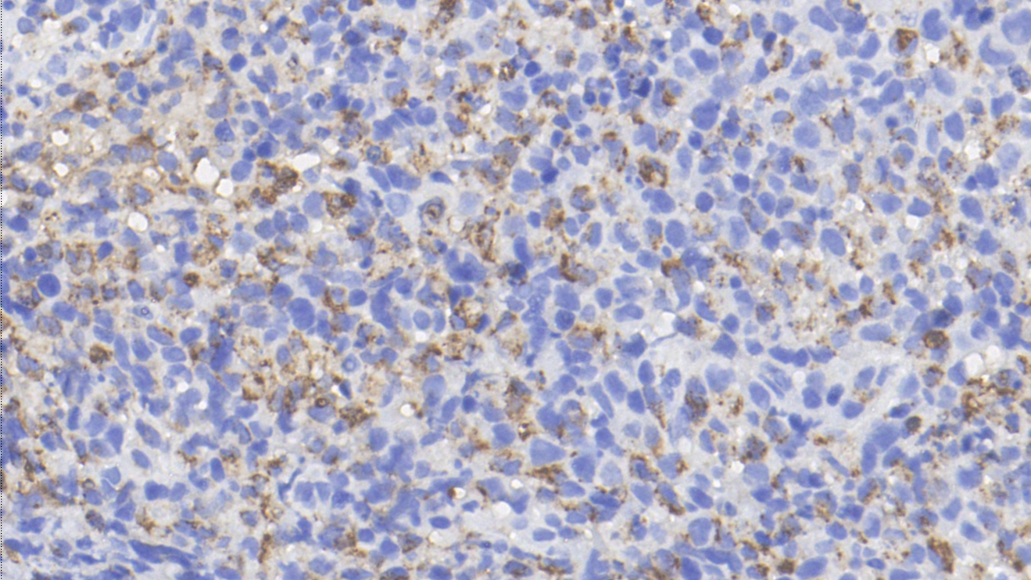
组织细胞:MPO少量+
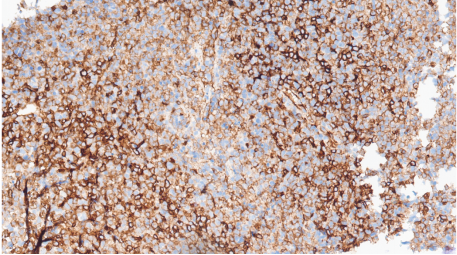
CD123部分+,强阳性细胞为浆细胞样树突状细胞
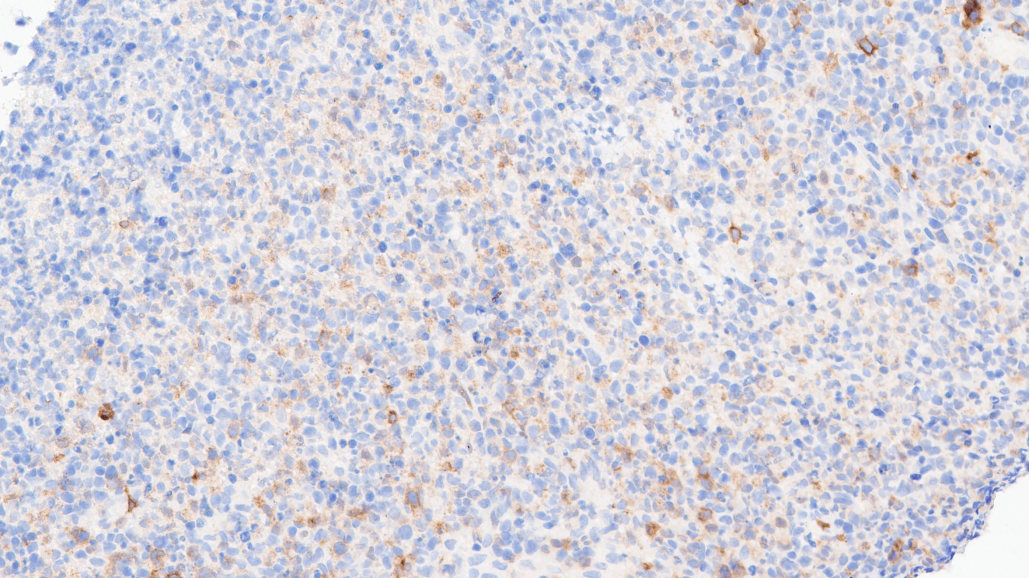
CD30散在+
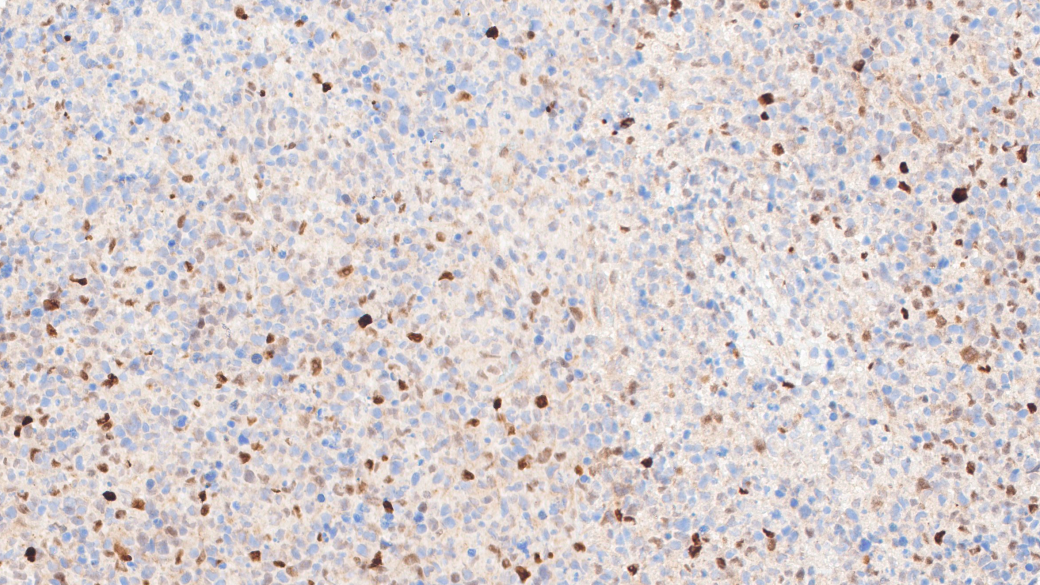
C-MYC散在+
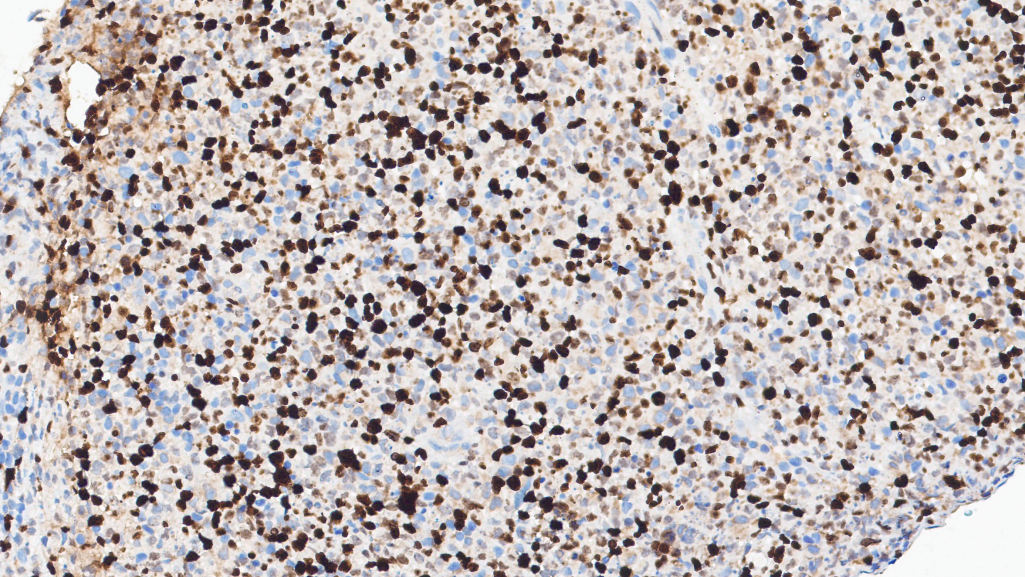
Ki67(热点区约60%+)
2、阴性标记:
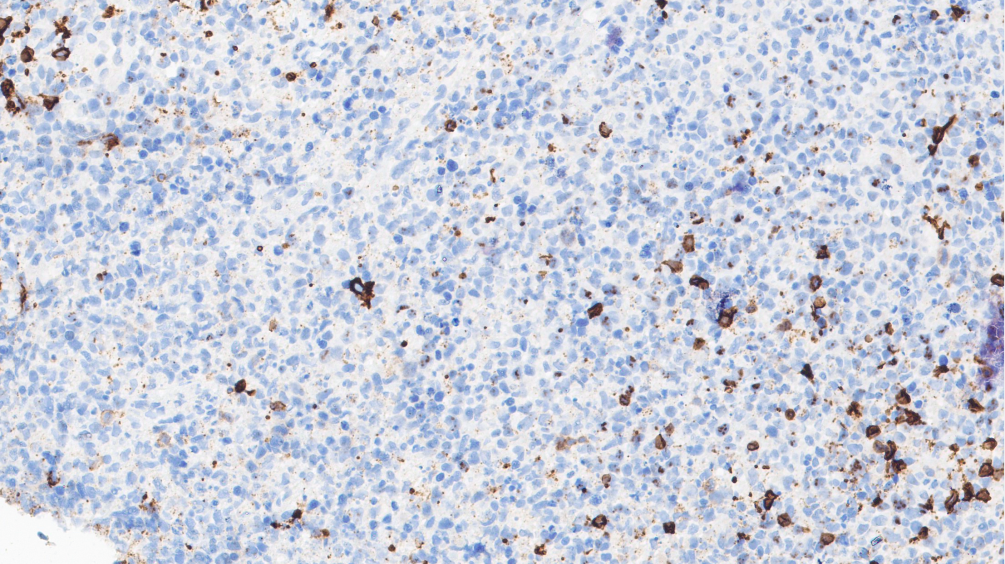
病灶区淋巴细胞:CD20-
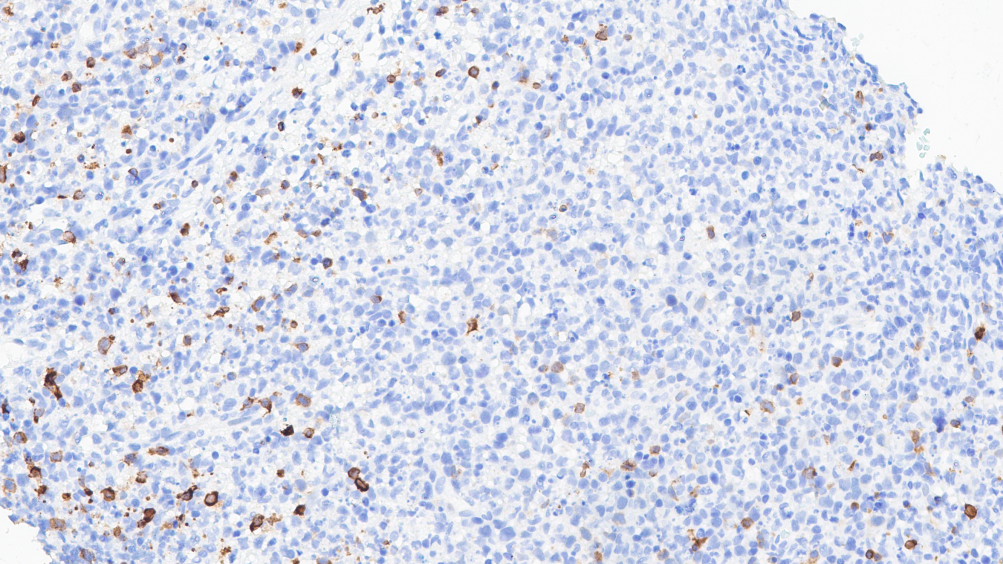
CD79a-
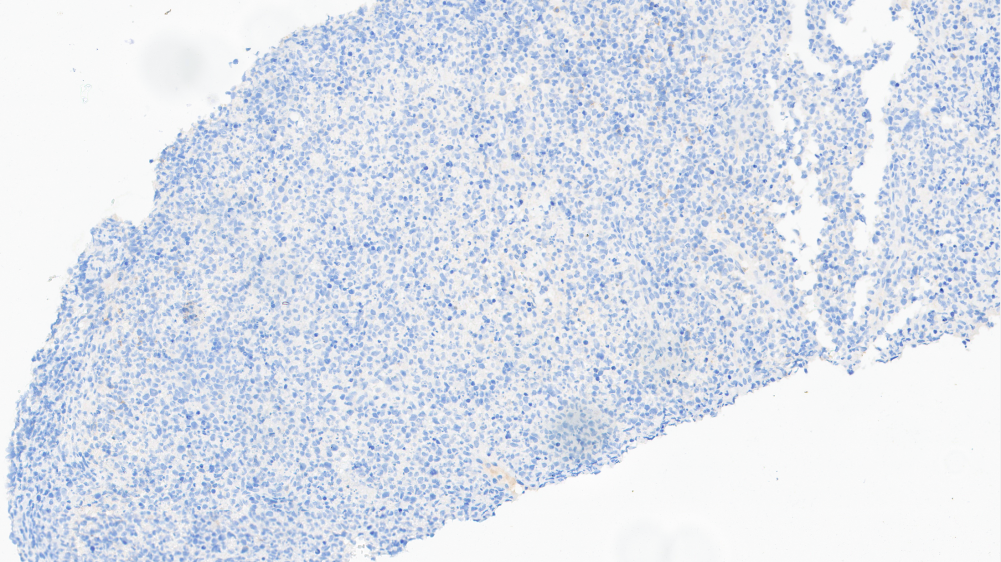
CD21-
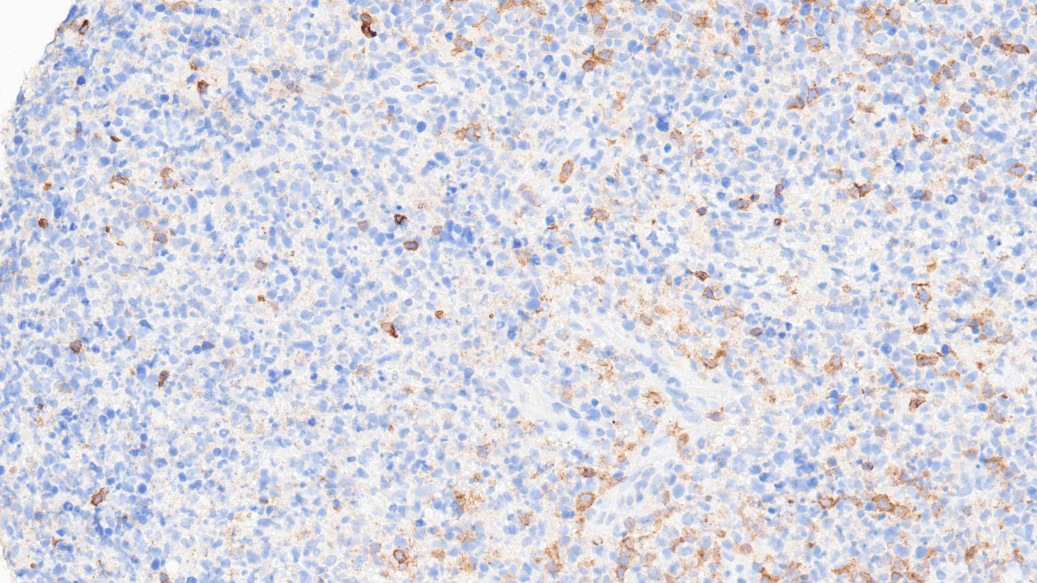
CD4病灶内增生淋巴细胞-,散在中等强度阳性细胞为组织细胞
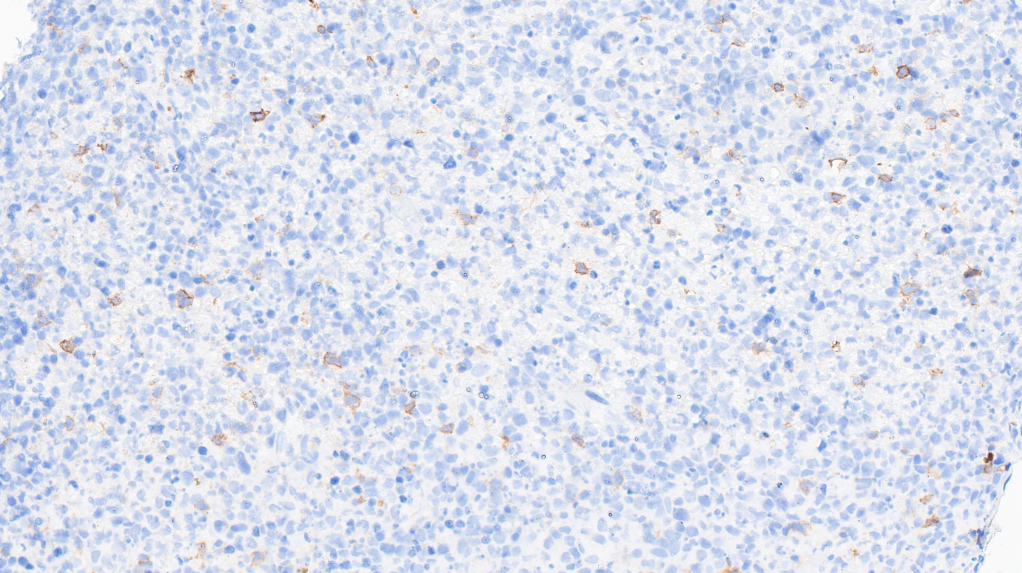
CD56-
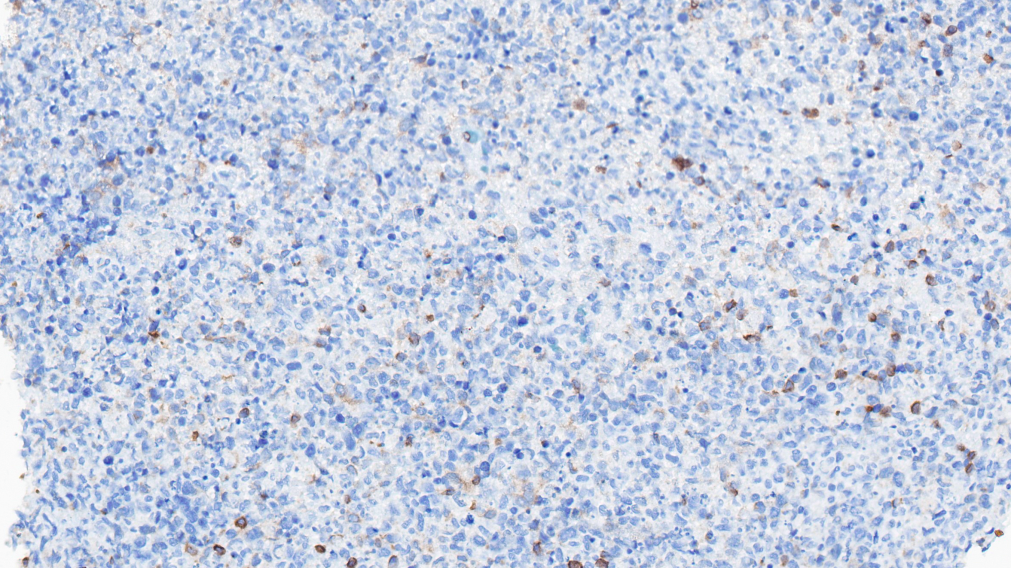
BCL-2-

BCL-6-
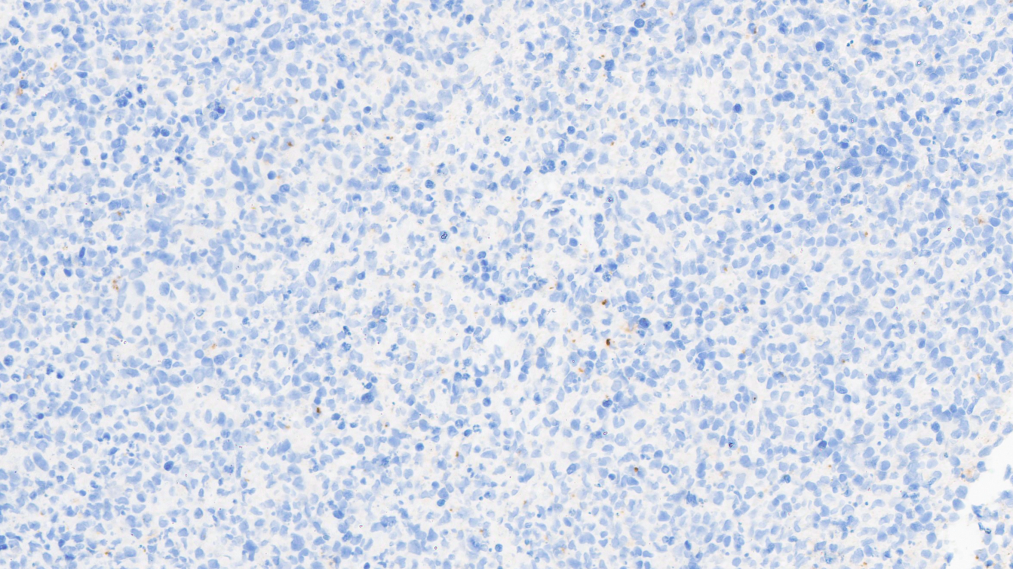
CD15-
EBV原位杂交:EBER-
免疫组化及原位杂交结果汇总:
阳性标记:
病灶区淋巴细胞:
CD3(+)、CD5(少量+,表达下调)、CD2(+)、CD7(+)、CD8(+)、TIA-1(+)、Granzyme-B(+)、Proforin(+)、C-MYC(5%-20%+)、MUM1(少量+)、CD30(个别+)、Ki67(热点区约60%+)
组织细胞:
CD68(+)、CD163(+)、MPO(少量+)
浆细胞样树突状细胞:
CD123(+)
阴性标记:
病灶区淋巴细胞:
CD20(-)、CD79a(-)、CD4(-)、Kappa(-)、Lambda(-)、CD56(-)、ALK(-)、CD10(-)、BCL-2(-)、BCL-6(-)、CD15(-);CD21(残存FDC网+)
EBV原位杂交:
EBER(-)
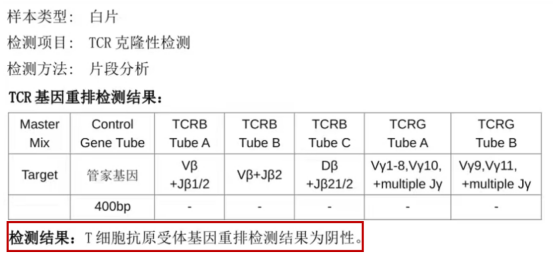
分子检测
最终诊断
“右颈部淋巴结穿刺活检标本”:Kikuchi–Fujimoto病(又称为组织细胞坏死性淋巴结炎)。
Kikuchi–Fujimoto病(组织细胞坏死性淋巴结炎)
定义
Kikuchi–Fujimoto disease (KFD)是一种自限性疾病。镜下特点为副皮质区免疫母细胞增生,具有特征性核的组织细胞浸润,以及细胞凋亡或伴大量核碎片的坏死。
第五版WHO淋巴造血系统肿瘤分类中,将其列为T细胞为主的肿瘤样病变。
临床特点
流行病学:多发生于年轻成人(中位年龄23-30岁),女性多见,但在儿童患者中男性多发。文献报道最大患病年龄为92岁。
主要症状:大部分KFD常累及单侧颈部淋巴结,也可全身多发淋巴结肿大。
有时仅累及肠系膜淋巴结,或表现为胸腔积液及中枢神经系统的症状。
影像表现:KFD因淋巴结多发肿大、坏死,结构不清、代谢增加等原因,使得影像学易误诊为恶性肿瘤。研究表明,病变区域的高代谢可能与疾病严重程度相关。
病理学-组织学
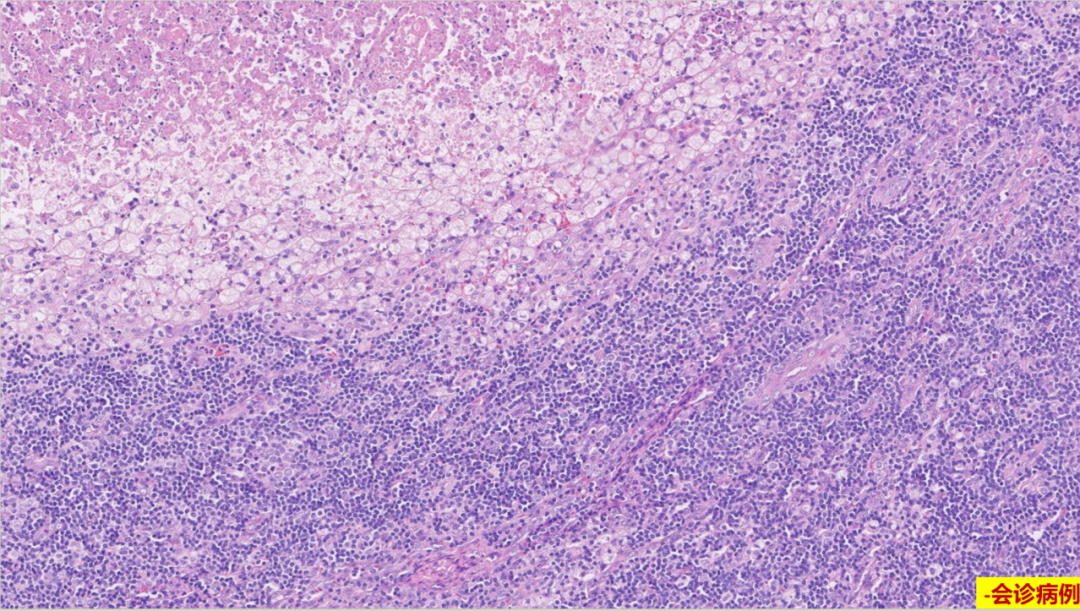
低倍镜特点:
不规则淡染坏死灶,多灶性,皮质、髓质均可发生,多个病灶融合,有时呈楔形。
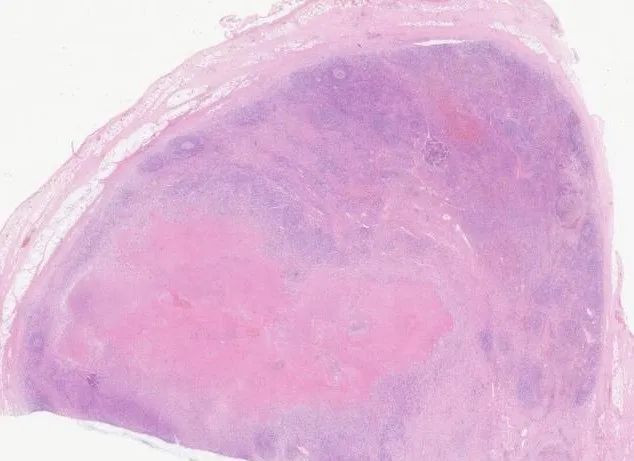
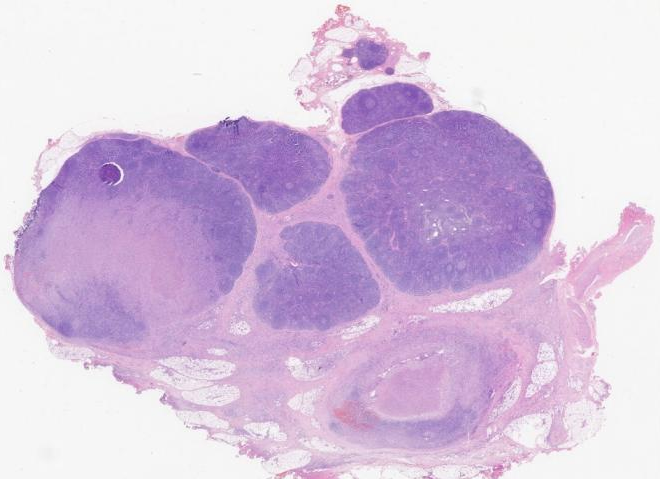
可见坏死灶毗邻有“星空”的副皮质区
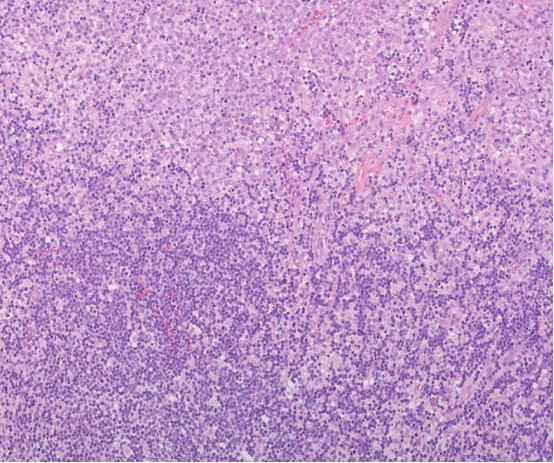
上方为坏死灶,下方为有“星空”的副皮质区
高倍镜特点:
HE形态上病灶区可分为三个期:增生期、坏死期、黄色瘤变期。
1.增生期:
大量核碎片、伴有扭曲或新月形核的组织细胞(常伴有吞噬核碎片现象)、免疫母细胞和浆细胞样树突状细胞。易误诊为淋巴瘤。
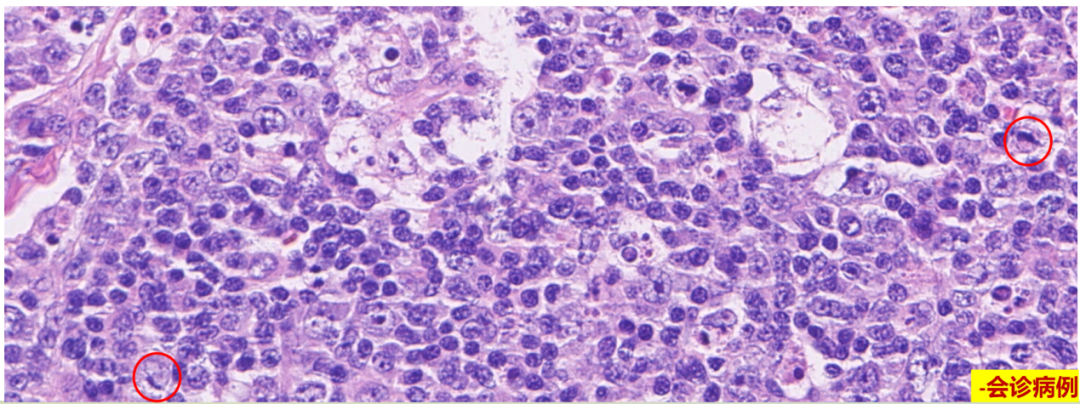
2.坏死期:
凝固性坏死,活跃的细胞崩解现象,无中性粒细胞浸润。
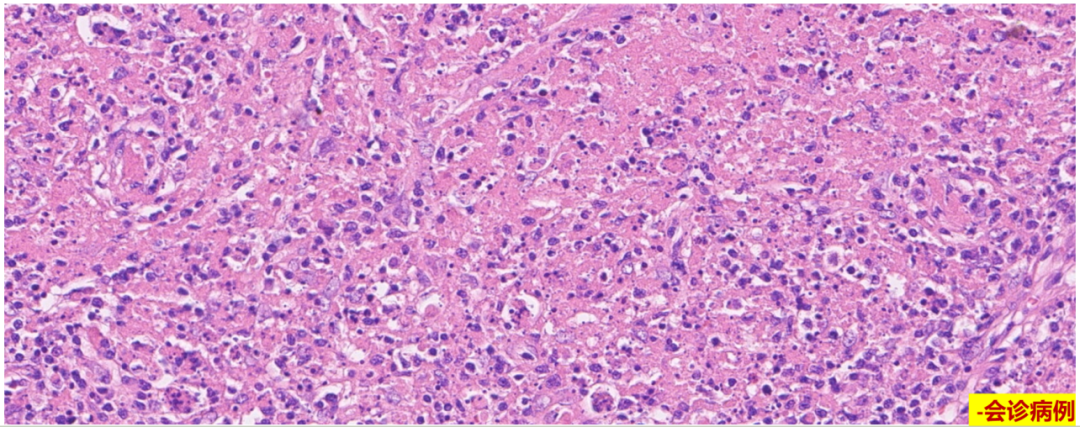
3.黄色瘤变期:
以成片增生的泡沫样组织细胞为特点,粒细胞缺如,浆细胞罕见。
免疫组化、EB病毒原位杂交及分子改变:
-
病灶区增生的免疫母细胞样淋巴细胞:CD3(+),CD2(+),CD7(常+),CD8(+),细胞毒性标记物(+),CD5常丢失或表达降低;CD20(-),CD79a(-),CD4(-),CD56(-);CD30(+/-)
-
组织细胞:CD68(+),CD163(+),MPO(+)
-
浆细胞样树突状细胞:CD3(-),CD4(+),CD123(+)
-
Ki67高表达
-
EBER(-)
-
TCR基因重排绝大部分为阴性,偶尔可阳性
诊断标准:
基本标准:
淋巴结受累;副皮质区散在分布核碎片,伴组织细胞、浆细胞样树突状细胞和免疫母细胞增生;无粒细胞,浆细胞罕见;并尽可能排除感染、淋巴瘤和自身免疫性疾病。
理想标准:
组织细胞具有新月形核并吞噬核碎片。
鉴别诊断:
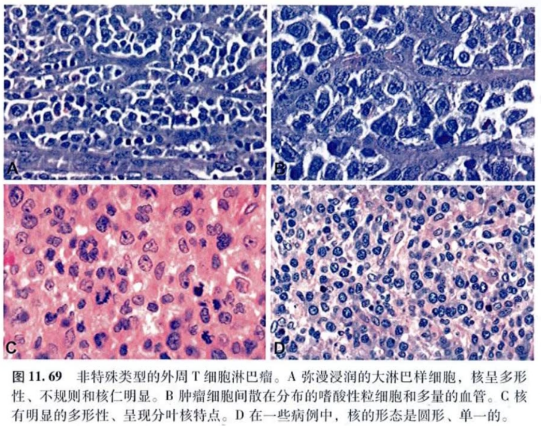
1.外周T细胞淋巴瘤(PTCL):
-
淋巴结呈膨胀性生长,大量多形性异型T淋巴细胞
-
可有粒细胞浸润,间质常伴血管增生
-
CD68阳性组织细胞少
-
TCR基因重排呈单克隆性
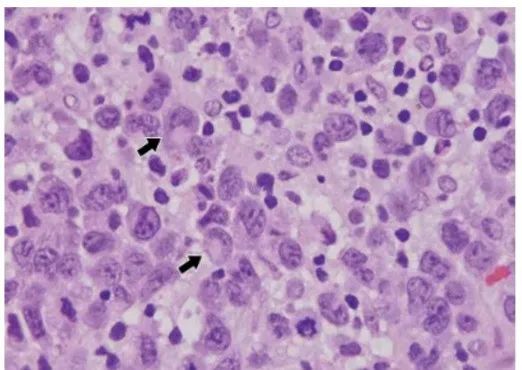
2.间变性大细胞淋巴瘤(ALCL):
-
增生淋巴细胞异型,并见核呈肾形或马蹄形淋巴细胞(hallmark cells)
-
CD30(弥漫+)
-
T细胞标记:CD2、CD3、CD4、CD5多数阳性
-
绝大多数TCR基因重排单克隆性
3.NK/T细胞淋巴瘤:
-
细胞异型,常可见核分裂象,伴坏死
-
可见血管中心性生长及浸润血管壁现象
-
CD3(+)、CD5(-)、CD56(+)、CD4和CD8常均-、细胞毒性颗粒(+)
-
EBER(+)
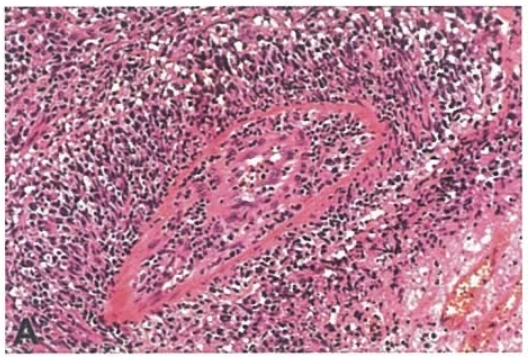
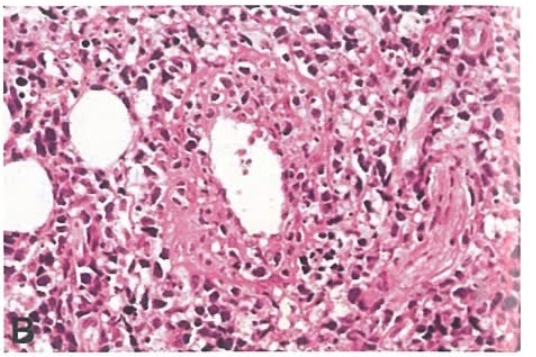
4.猫抓病(CSD)
-
临床表现:儿童和青少年,小动物抓伤或咬伤病史;皮肤红斑、丘疹、水疱、结痂;1-3周后淋巴结肿大
-
病理学:坏死区呈不规则形和星形,伴大量中性粒细胞,周边为栅栏排列的组织细胞;淋巴窦内充满单核细胞样B淋巴细胞
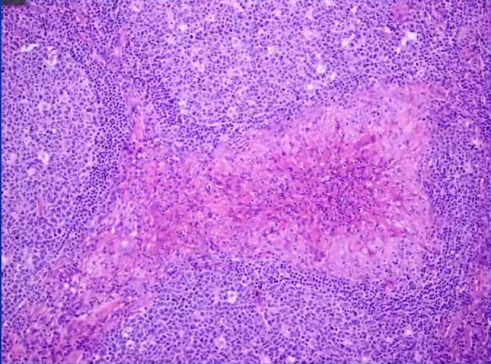
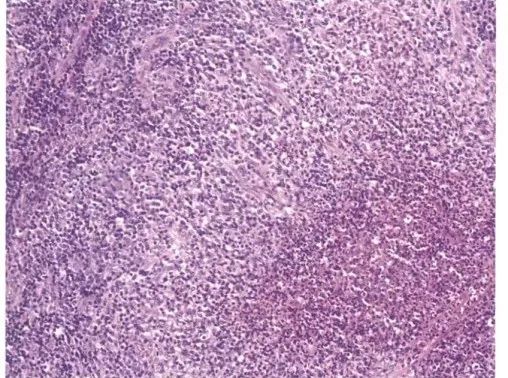
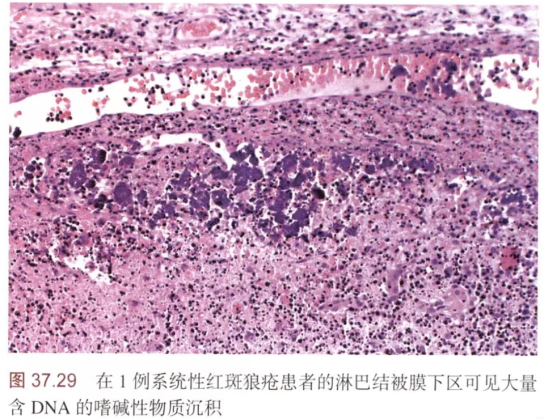
5.系统性红斑狼疮(SLE)
-
临床表现:颧部红斑、盘形斑、口腔溃疡、关节炎、光过敏等
-
病理学:可见苏木素小体,中性粒细胞浸润,血管炎
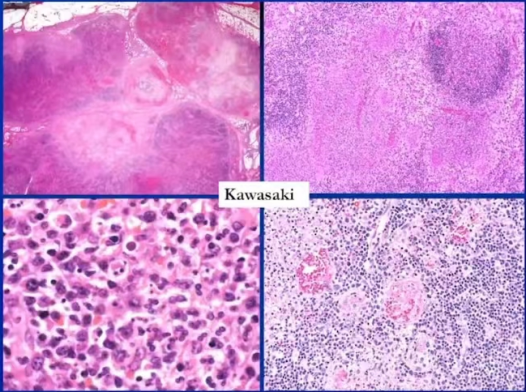
6.皮肤黏膜淋巴综合征(Kawasaki)
-
临床表现:主要累及5岁以下亚洲儿童
临床进程分二期:
第一期:高热至少持续5天以上
第二期:脱皮(手、足多见);关节痛;腹泻、腹痛;呕吐
-
病理学:地图样纤维素性坏死,伴中性粒细胞浸润;未见浆细胞样树突状细胞;纤维素性血栓;动脉炎
治疗及预后
-
治疗:目前无统一的治疗方案,糖皮质激素治疗可明显缩短病程。
-
预后:儿童复发率为12.2%,1%的患者可发展为系统性红斑狼疮,而在成人中,KFD复发与抗核抗体阳性有关。建议随诊、监测自身免疫性疾病的发展。
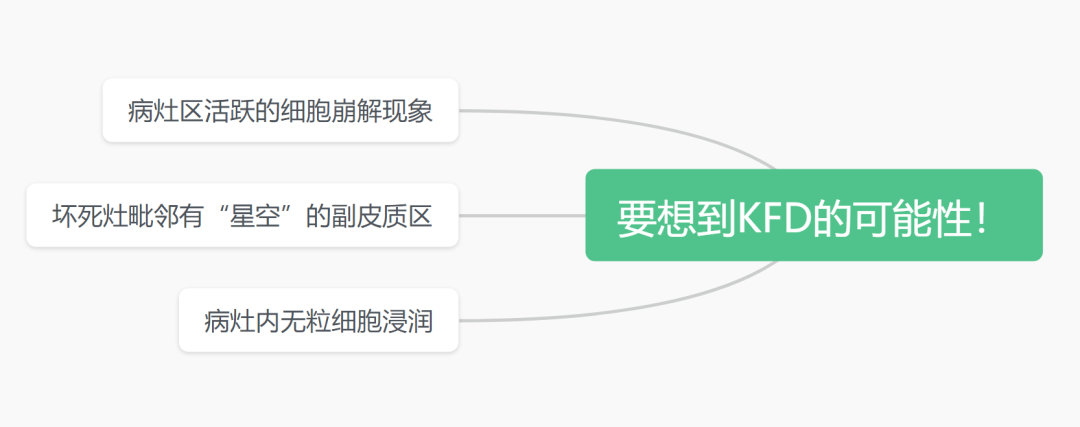
小结
KFD诊断的形态学线索(经验分享)
参考文献及书籍:
1.The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours:Lymphoid Neoplasma.
2.Rosai and Ackerman’s Surgical Pathology.
