Genome Res:变分图卷积自编码器框架——PAST,可融合外部参考数据进行空间转录组数据降维分析
时间:2023-11-18 21:37:28 热度:37.1℃ 作者:网络
近日,清华大学自动化系江瑞课题组和南开大学数学科学学院陈盛泉课题组在Genome Research上发表了题为“Latent feature extraction with a prior-based self-attention framework for spatial transcriptomics”的论文,提出了融合生物先验信息和空间位置信息来解析空间转录组(Spatial Transcriptomics)数据、提取低维表示特征的变分图卷积自编码器框架PAST。该模型不仅可以有效提取空间转录组数据的低维表示特征,辨识空间功能区域(Spatial Domains),还能够促进数据可视化、发育轨迹推断和伪时序分析等多种下游任务。此外,PAST还能够进行多切片共嵌入降维分析,进而完成数据整合与标签迁移,促进空间区域自动注释。

文章发表在Genome Research
空间转录组数据解析是目前生物信息学的研究前沿与热点。空间转录组测序技术被Nature Methods杂志评为2020年度方法,也被誉为继单细胞转录组测序技术之后进行组织生物学研究的下一代测序技术。鉴于目前转录组数据的大量积累,已有的空间转录组数据分析算法都没有考虑到利用已有生物数据中蕴含的丰富先验信息,而且同时融合生物先验信息、空间位置信息和基因表达信息也对现有方法的跨模态信息融合能力提出了挑战。此外,空间转录组测序通量不断增加,这也对现有方法在大规模数据集上的可拓展性提出更高的要求。
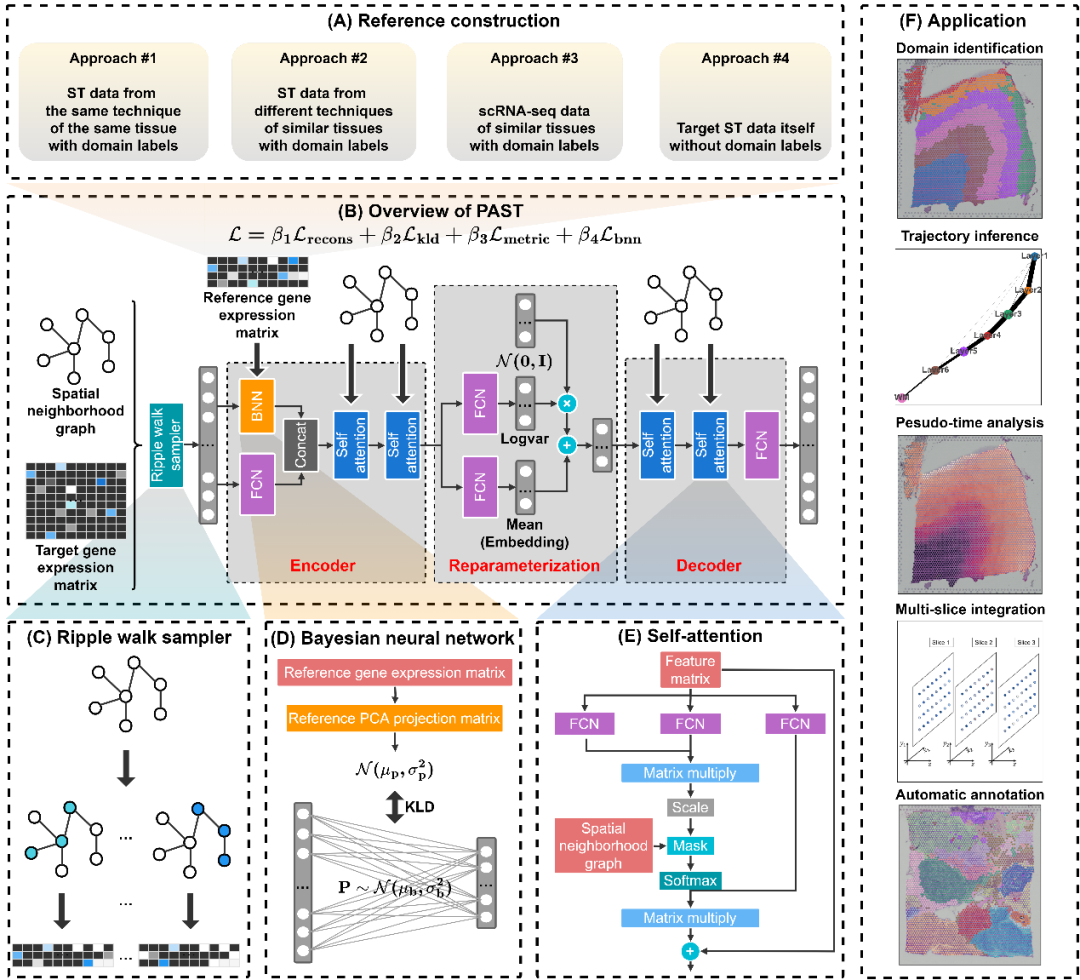
图1. PAST模型示意图
针对上述难点,江瑞课题组与陈盛泉课题组提出了PAST模型。如图1所示,PAST模型基于贝叶斯神经网络融合外部参考数据中的生物先验信息,基于图卷积神经网络和自注意力机制融合空间位置信息,基于波纹行走策略进行子图训练以提高拓展性,从而具有极强的信息融合能力和在大规模数据上的可拓展性。PAST可以融合来自多种来源数据的生物先验,进而得到空间转录组数据的低维表示特征,不仅在空间聚类、空间区域辨识等任务的性能上优于现有方法,还能有效促进空间可视化、发育轨迹推断和伪时序分析等下游分析(图2)。
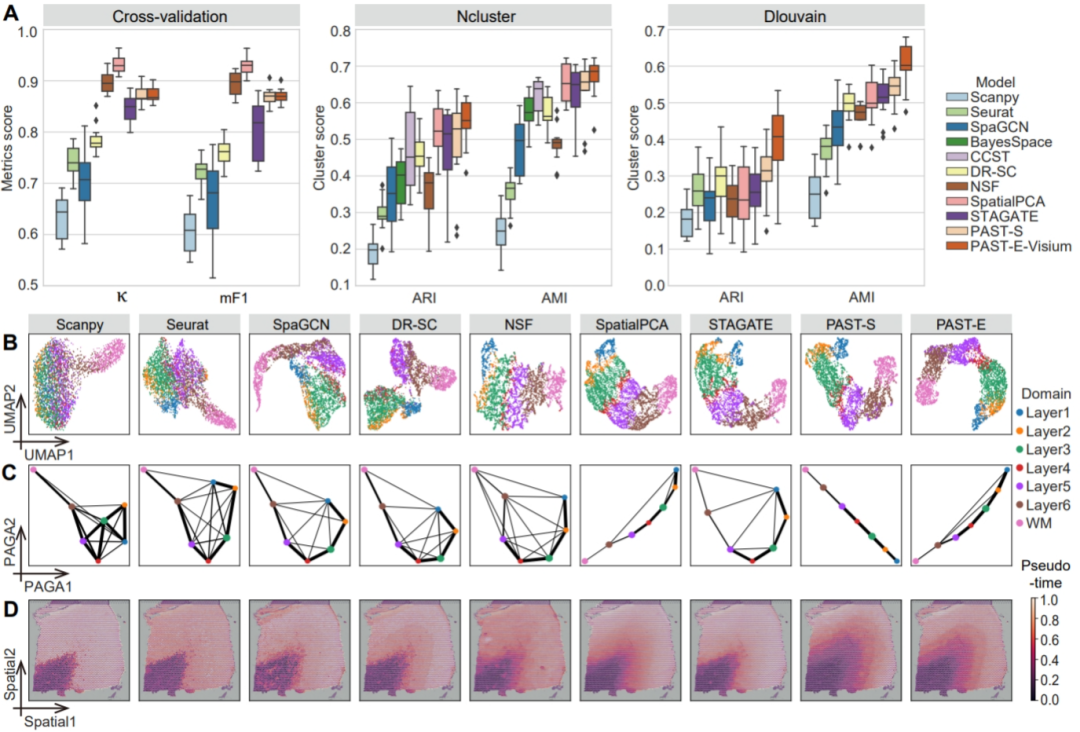
图2. PAST有效促进各种下游分析
PAST还能够进一步拓展应用于多切片数据整合与多切片标签迁移。基于所提出的迁移学习策略(PAST-TL)与三维堆叠策略(PAST-3DStack),PAST能够对多张切片进行共嵌入降维分析,得到多张切片在同一隐空间的联合低维表示,从而进行低维可视化和标签迁移等分析。在人类背外侧前额叶皮质数据集和人类乳腺癌数据集数据集上,PAST的多切片联合低维表示不仅可以捕获生物差异,还不受到切片间批次效应的影响(图3A, B, E, F)。此外,基于PAST模型所得到的联合低维表示也能获得准确的标签迁移结果,有效促进跨切片空间功能区域自动注释(图3C, D, G)。
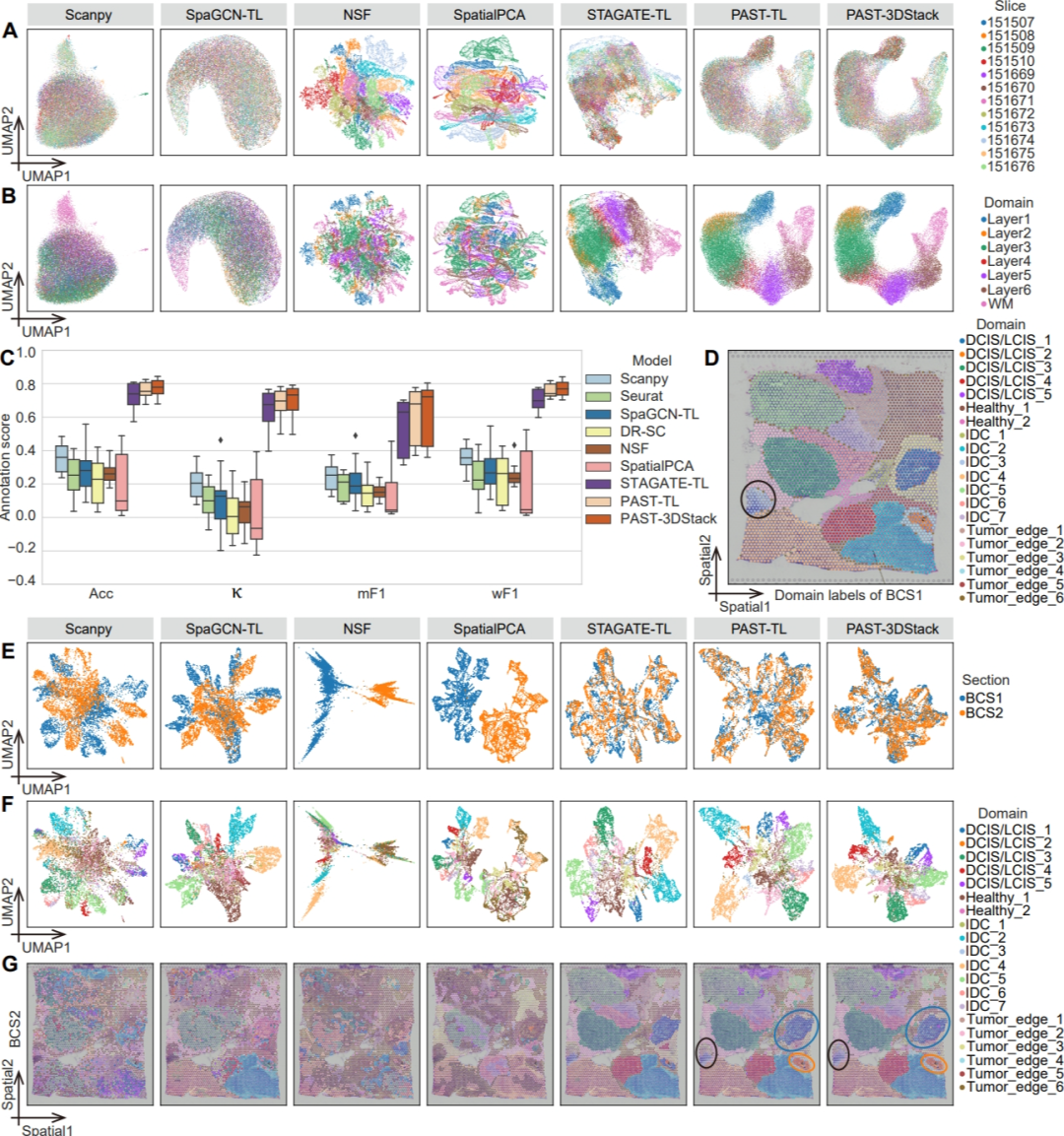
图3. PAST有效促进多切片整合与标签迁移
综上所述,PAST通过融合多种来源的生物先验信息,能够有效促进空间转录组数据的各种下游分析任务,并完成多切片数据整合与标签迁移任务,有望成为空间转录组数据分析的重要工具。
清华大学自动化系2022级直博生李震为该论文的第一作者,清华大学自动化系2020级直博生陈晓阳、清华大学自动化系张学工教授和清华大学自动化系江瑞教授为本研究做出了重要贡献,南开大学数学科学学院信息与数据科学系陈盛泉副教授为本文的通讯作者。该课题得到了科技部重点研发计划、国家自然科学基金委等的资助。
原文链接:
https://genome.cshlp.org/content/early/2023/10/30/gr.277891.123.abstract

