肿瘤抑制因子p53最初被认为是癌基因,但实际上野生型p53(wtp53)是一种真正的肿瘤抑制因子,突变体p53(mutp53)的功能作为独立于野生型p53(wtp53)的癌基因。
p53是人类癌症中最常见的突变基因,在超过50%的人类癌症中发生突变。鉴于p53的突变通常在肿瘤中特异性观察到,并且在非肿瘤组织中很少见,因此mutp53是理想用于癌症治疗的治疗靶点。
国内外药企纷纷布局p53,然而历经40多年的研究开发,人们仍然对这些突变的p53蛋白束手无策,至今仍未有p53药物获批上市,罗氏、强生、诺华等大药企纷纷折戟于此,强生、默沙东也先后宣布暂停相关的研究项目。
p53蛋白的表面光滑,没有合适的口袋能够与化合物结合,在成药方面有着巨大的挑战,这也使得p53成为与RAS、MYC齐名的三大不可成药靶点之一。
目前临床上采取了几种靶向p53突变的策略,包括直接靶向错义mutp53,使mutp53蛋白的消耗和降解或诱导p53合成致死性或靶向p53突变或缺失而导致增加的酶等。
除此之外,人们也在开发基于p53免疫治疗,包括基于p53的疫苗,p53特异性抗体和基于p53的基因治疗等。
继RAS成为可靶向的靶点后,相信不久的将来mutp53也一定会成为可成药的靶点。
肿瘤抑制因子p53最初被认为是癌基因,因为最初克隆的cDNA含有错义突变。事实上,突变体p53(mutp53)的功能作为独立于野生型p53(wtp53)的癌基因。在使用没有任何突变的p53 cDNA的研究中,wtp53被证明是一种真正的肿瘤抑制因子[1]。
p53是一种转录因子,可调节参与细胞凋亡、细胞周期停滞、衰老、DNA修复和细胞代谢的许多下游靶基因的转录,从而起到肿瘤抑制作用[2]。
蛋白质水平和活性的wtp53在非应力条件下保持较低水平,主要是通过MDM2降解。
在遗传毒性条件下,wtp53通过磷酸化或乙酰化的翻译后修饰(PTM)稳定和激活,以诱导细胞周期停滞和/或细胞死亡(图1)[3]。
一旦wtp53功能因突变或缺失而受损,细胞就会失去对其生长的控制,从而促进肿瘤发生。越来越多的证据表明,p53是人类癌症中最常见的突变基因,在超过50%的人类癌症中发生突变。大多数p53突变是DNA结合域中的错义突变。
p53突变导致作为转录因子和肿瘤抑制因子的功能丧失(LOS)。错义mutp53 经常在肿瘤中积聚,以独立于wtp53的方式促进恶性进展、转移和耐药性。这些致癌性mutp53活性被称为功能获得(GOF)(图1)。
mutp53 GOF的机理主要是由mutp53与肿瘤抑制因子结合的能力引起的(例如,p63,p73,MRN复合物)和癌基因(例如,ETS2,SREBP2,NF-Y)以改变这些结合伴侣的功能。
临床上,肿瘤中mutp53的存在与多种类型癌症患者的晚期临床分期、转移和不良结局密切相关。鉴于p53的突变通常在肿瘤中特异性观察到,并且在非肿瘤组织中很少见,因此mutp53是理想用于癌症治疗的治疗靶点。
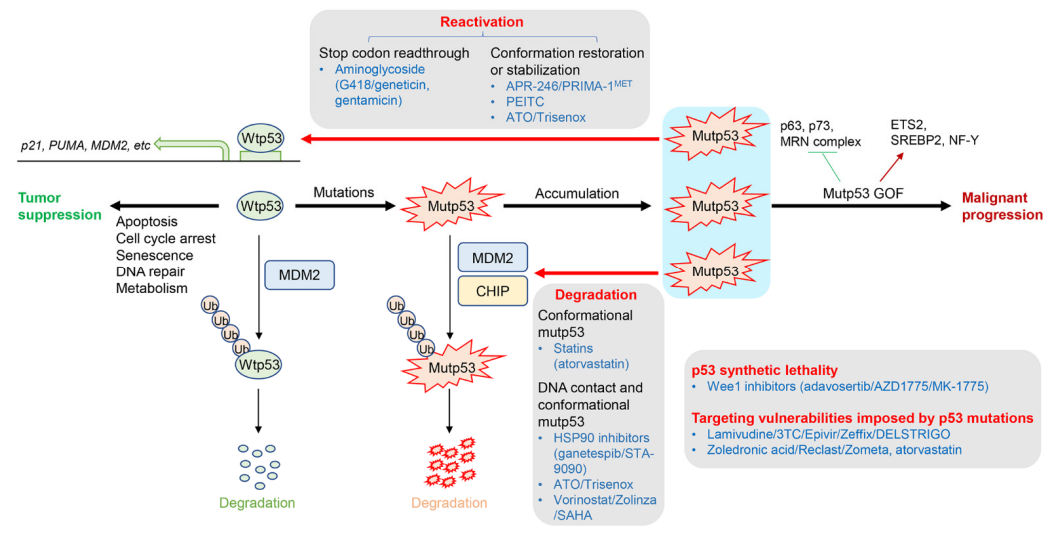
图1. wtp53、mutp53 GOF的功能以及靶向p53突变的策略
直接靶向错义mutp53,以恢复wtp53的活性或稳定wtp53构象
大多数mutp53失去wtp53作为转录因子和肿瘤抑制因子的活性。越来越多的证据表明,肿瘤抑制活性可以在特定条件下恢复,包括温度变化、暴露于来自53BP2 衍生的“CDB3”和p53 C端结构域“肽 46”的合成肽,以及插入第二位点突变或 N 末端缺失[4,5]。
人们试图发现恢复wtp53构象、转录活性和肿瘤抑制活性的小分子化合物。例如,CP-31398 是最早能稳定p53活性确认的mutp53再活化化合物并促进p53介导的肿瘤抑制。此外,氨基苯并噻唑类似物JC744最近被鉴定为一种新化合物,可以通过p53Y220C诱导的窄表面袋在体外特异性结合并稳定p53Y220C突变体。
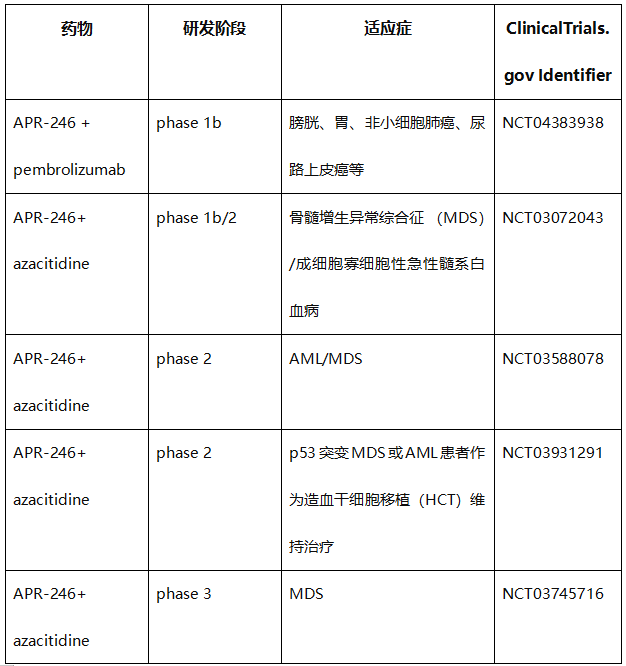
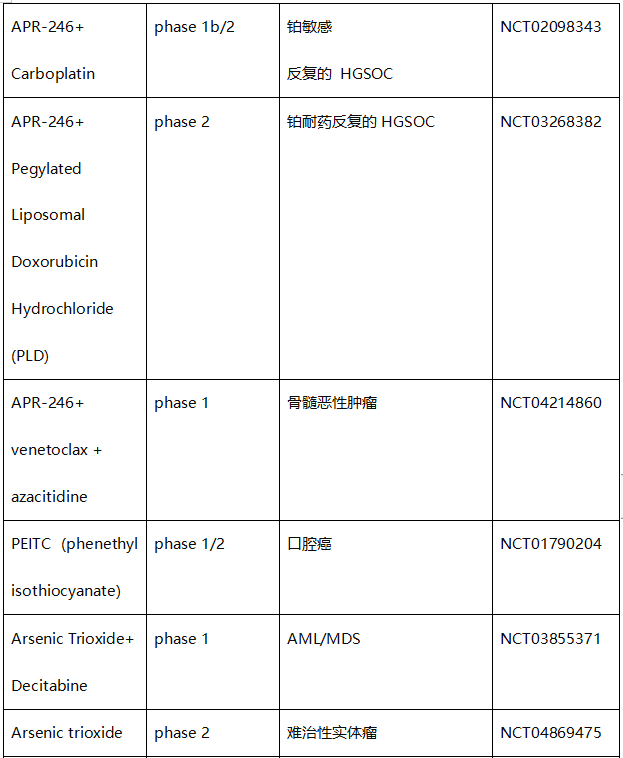
然而,只有少数化合物处于临床试验状态。这些包括APR-246(eprenetapopt/PRIMA-1MET)、异硫氰酸苯乙酯(PEITC)和三氧化二砷(ATO/Trisenox)(表1)。从表1可以看出,APR-246在临床上开展的试验都是联合治疗,目前还没有开展APR-246单药给药的临床试验。
表1. 直接靶向错义mutp53的化合物的临床试验
APR-246(Eprenetapopt,PRIMA-1MET)是由Aprea Therapeutics公司开发的,能够恢复突变型p53的活性,并诱导凋亡。APR-246是一种前体药物,能够转化为活性化合物methylene quinuclidinone(MQ, 一种Michael 受体,通过半胱氨酸与突变型p53结合并恢复p53野生型构象)。
在耐药性卵巢癌细胞中,APR-246能够完全恢复p53突变体对cisplatin和doxorubicin的敏感性。它不仅重新激活p53,也能以剂量依赖方式降低细胞内谷胱甘肽的水平。
APR-246通过诱导ROS、ER胁迫和抑制TrxR1,引起p53依赖性细胞凋亡。在骨髓癌细胞中,APR-246通过破坏GSH/ROS的平衡,不受p53的影响下,诱导细胞凋亡。PRIMA-1Met/APR-246在体内能够有效地抑制表达突变型p53的SCLC细胞的生长并诱导期凋亡,DNA片段化增多、caspase-3激活、PARP分裂、Bax和Noxa上调、Bcl-2下调。
APR-246在骨髓增生异常综合征(MDS)适应症上已获FDA认定突破性疗法、孤儿药地位和快速通道资格,急性髓系白血病(AML)适应症获FDA快速通道资格认定,欧洲药品管理局给予其治疗MDS、AML和卵巢癌的孤儿药地位。
然而,2020年12月28日, Aprea Therapeutics宣布APR-246联合阿扎胞苷一线治疗骨髓增生异常综合征(MDS)的III期临床未达主要临床终点。该消息使Aprea Therapeutics的股价瞬间崩盘,下跌78%,由前一日的25.09美元/股降至28日收盘的5.50美元/股。
越来越多的证据表明,mutp53本质上是不稳定的。遗传毒性应激,包括致癌应激和辐射,稳定mutp53以促进其在肿瘤中的GOF活性。至关重要的是,敲低mutp53可减少恶性癌细胞的特性,表明癌细胞对致癌性mutp53成瘾。mutp53稳定或降解的确切机制尚不清楚,但mutp53特异性降解机制与wtp53不同。
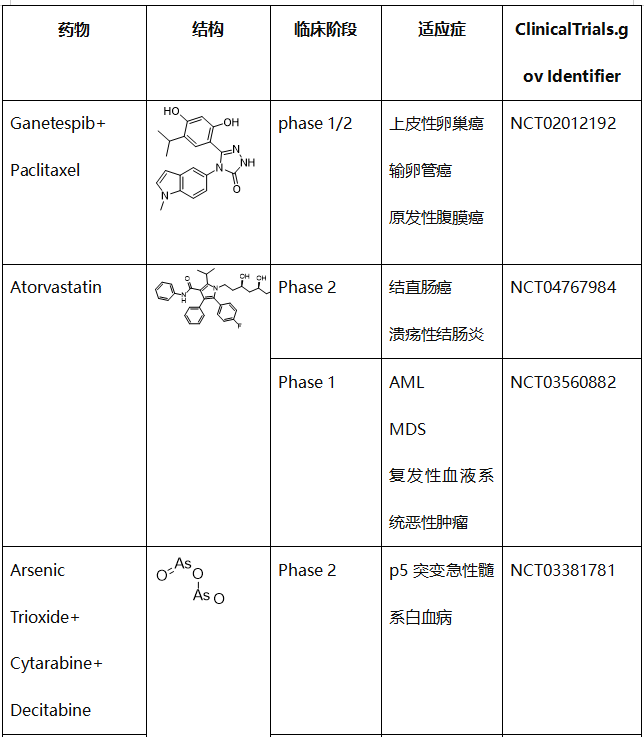
事实上,有几种化合物在不改变wtp53水平的情况下诱导mutp53降解,一些降解mutp53的药物正在临床试验中。这些药物包括 HSP90 抑制剂(ganetespib/STA-9090)、他汀类药物(阿托伐他汀)、ATO/曲森诺和伏立诺他(SAHA)(表2)。
表2. 使mutp53蛋白的消耗或降解的化合物的临床试验
HSP90是一种ATP酶依赖性分子伴侣,可调节蛋白质折叠和在生理和压力条件下稳定或降解蛋白质,从而控制蛋白质平衡。HSP90也显示与wtp53和mutp53结合。
HSP90调节wtp53在生理和非生理条件下的活性。HSP90还通过形成复合物来抑制MDM2和CHIP泛素连接酶,从而有助于mutp53的稳定。因此,HSP90在wtp53和GOF mutp53活性或蛋白质稳定性中的作用是多种多样的;多样性可能取决于细胞环境、压力类型和客户结构。
Ganetespib(STA-9090)是一种合成的HSP90间苯二酚三唑啉酮抑制剂,正在临床试验中。Ganetespib与HSP90的N末端的ATP结合结构域结合,灭活或下调致癌HSP90,导致肿瘤抑制。
诱导p53合成致死性或靶向p53突变或缺失而导致增加的酶
合成致死性是诱导细胞死亡的生物过程,其基于同时抑制在细胞存活所需的过程中的两种途径并行作用,仅抑制一种途径会导致细胞存活。合成致死策略已在抗癌治疗中得到广泛应用。
一些具有p53缺失或突变的合成致死伴侣已有报道(例如,Wee1、PKC、PLK1、PARP),其中许多在G2和M细胞周期检查点中起作用。考虑到p53的主要功能是调节G1细胞周期检查点,损害对维持DNA复制和基因组或染色体完整性至关重要的其他细胞周期检查点(G2,M)可能对细胞有害。
1)由wee1抑制剂adavosertib诱导p53的合成致死性
目前,只有 Wee1 抑制剂adavosertib(AZD1775/MK1775) 正在临床试验中,以检查它是否有效抑制具有p53缺失或突变(p53缺乏症)的肿瘤进展。
Adavosertib已在总共59项临床试验中进行了测试。其中,已经进行了九项2期试验,对癌症中的p53状态进行分层,包括卵巢癌(NCT01164995,NCT01357161,NCT02272790),复发性SCLC(NCT02688907,NCT02593019),未经治疗的非小细胞肺癌(NSCLC,NCT02087241),NSCLC (NCT02087176)、复发性子宫浆液性癌 (NCT03668340) 和结直肠癌 (FOCUS4-C)。
具体而言,在NCT01164995试验中,adavosertib增强了卡铂对p53突变卵巢癌的疗效,总缓解率43%。此外,在NCT01357161试验中,adavosertib可提高卡铂/紫杉醇化疗对有p53突变的铂类敏感卵巢癌女性的治疗效果,通过改善PFS(7.9个月vs7.3个月;风险比:HR,0.63),观察到adavosertib具有适度的临床获益。
此外,在NCT02272790试验中,调查了adavosertib与常用化疗药物(吉西他滨、紫杉醇、卡铂或聚乙二醇化脂质体阿霉素)联合使用对于原发性铂类耐药卵巢癌的有效性和安全性,adavosertib与卡泊拉联合治疗显示出一些有希望的结果。然而,这种联合治疗比卡铂单药治疗更频繁地引起血液学毒性。因此,未来需要对adavosertib和卡铂组合优化治疗剂量和方案的研究。
2)靶向因p53缺乏而增强的LINE-1的逆转录酶
LINE-1(长穿插元件1)是存在于人类基因组中的反转录转座子元件家族,由~15%的基因组组成。LINE-1的迁移性在体细胞组织中在很大程度上受到抑制,但在许多癌症中被抑制,其中LINE-1逆转录转位与p53突变和拷贝数变更(CNA)相关。
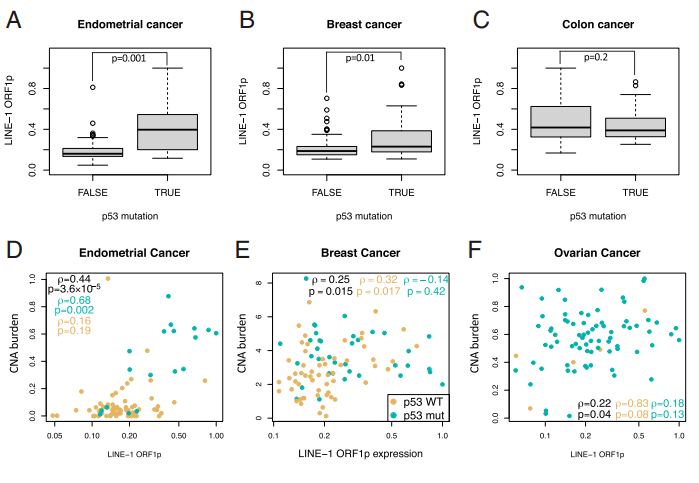
越来越多的报道显示wtp53功能丧失或p53突变与LINE-1表达相关,而wtp53通过与5‘UTR内的L1内部 RNA 聚合酶II启动子结合来抑制LINE-1表达(图2)。这些发现表明,在p53缺陷的癌症中被抑制的LINE-1导致基因组不稳定和恶性肿瘤进展[6]。
图2. A-C. 分别比较子宫内膜、乳腺癌和结肠肿瘤中 p53 野生型与突变肿瘤中 LINE-1 表达的箱形图;D–F. 子宫内膜癌、乳腺癌和卵巢癌中 LINE-1 表达与 CNA 负荷之间的 Spearman 相关性
事实上,FDA批准的HIV-1非核苷RT抑制剂依非韦伦抑制LINE-1 ORF2p中编码的逆转录酶(RT)活性可减少乳腺癌细胞和胰腺癌细胞的增殖Lamivudine(3TC/Epivir/Zeffix/DELSTRIGO)是FDA批准的用于治疗HIV和HBV的合成核苷RT抑制剂,可特异性降低人LINE-1逆转录转位(图3)[7]。
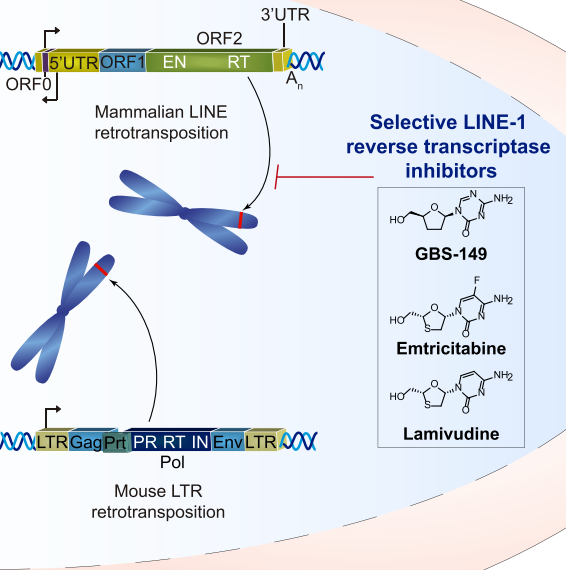
图3. Lamivudine抑制LINE-1
Rajurkar等人已经启动了单药Lamivudine在表达mutp53的化疗难治性转移结直肠癌患者中的2期临床试验(NCT03144804)。在参加本研究的 32 名患者中,有 8 名患者使用单药Lamivudine,观察到稳定疾病(SD)。
基线LINE-1 ORF1p的分析治疗前肿瘤活检和患者血清标本中的蛋白质水平显示进展性疾病(PD)组的ORF1p水平明显高于SD组,表明可能需要更高剂量的Lamivudine来抑制PD组中LINE-1的活性。尽管如此,他们的研究鼓励使用Lamivudine抑制由p53突变或缺失引起的LINE-1活性,这可能对癌症进展至关重要。
3)靶向使GOF mutp53增强的YPA/TAZ
越来越多的证据表明,GOF mutp53在功能上增强了YAP/TAZ活性,这极大地促进了癌症的进展。例如,几个DNA接触和结构GOF p53突变体与YAP相互作用,与致癌转录因子NF-Y形成复合物并增强转录活性(图4)[8]。
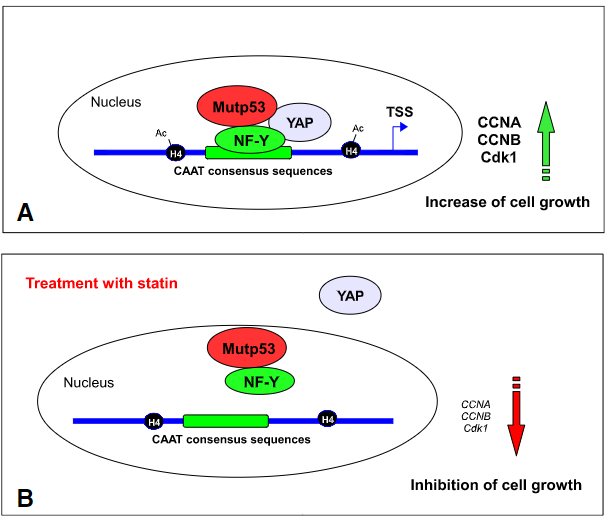
图4. mutp53与YAP/TAZ在细胞中作用
此外,mutp53结合并激活SREBP2,SREBP2是控制甲羟戊酸途径中酶表达的关键转录因子,导致异戊烯化增加RhoA,通过LATS1/2抑制YAP磷酸化,并增强YAP/TAZ的核易位和活化。因此,通过mutp53激活甲羟戊酸途径增强YAP/TAZ活性可能在表达MUTP53的癌细胞的恶性进展中起关键作用。由于对甲羟戊酸途径的抑制也会导致mutp53的蛋白水平降低,因此该途径的阻断可能导致抑制mutp53和YAP/TAZ的活性。
有FDA批准的药物抑制甲羟戊酸途径。一种是称为他汀类药物的降胆固醇药物,其抑制HMG-CoA还原酶。另一种是唑来膦酸(Reclast/Zometa),这是最常用的双膦酸盐之一,可抑制破骨细胞活性,因此用于治疗高钙血症、骨质疏松症、佩吉特病、多发性骨髓瘤和骨癌转移。
事实上,Sorrentino等人表明,色伐他汀或唑来膦酸在多种癌症类型中抑制YAP / TAZ的核定位和活性。
Göbel等人还报道了他汀类药物(辛伐他汀,瑞舒伐他汀,阿托伐他汀)与唑来膦酸的组合对诱导乳腺癌和前列腺癌细胞凋亡和毒性。
正在进行一项2期临床试验(NCT03358017),以检查阿托伐他汀和唑来膦酸联合使用标准蒽环类/紫杉烷类新辅助化疗治疗三阴性乳腺癌。这项试验不仅研究治疗对 YAP/TAZ 活性的变化,但也评估与新辅助标准相关的甲羟戊酸通路阻断的抗肿瘤作用通过对 p53 蛋白水平分层进行化疗(低 ≈ wtp53 与高 ≈ mutp53)。
总之,使用FDA批准的阻断甲羟戊酸途径的药物可以通过靶向两种致癌因子mutp53和YAP / TAZ的活性来抑制癌症进展。
p53中的突变具有癌症特异性,因此是靶向癌症治疗的理想分子靶标。几种药物已被开发用于靶向 p53突变,包括直接靶向错义mutp53,使mutp53蛋白的消耗和降解或诱导p53合成致死性或靶向p53突变或缺失而导致增加的酶等,但是目前还没有靶向mutp53的药物被批准上市。
除了开发小分子药物外,人们也在开发基于p53免疫治疗,包括基于p53的疫苗,p53特异性抗体和基于p53的基因治疗等。
mutp53蛋白因为表面光滑,没有合适的口袋能够与化合物结合,在成药方面有着巨大的挑战,这也使得p53成为与RAS、MYC齐名的三大不可成药靶点之一。
但是目前RAS已经有两款药物上市,打破它的“不可成药性”观点,相信不久的将来mutp53也一定会成为可成药的靶点。
参考文献:(上下滑动查看更多)
1.Zambetti, G.P.; Levine, A.J. A comparison of the biological activities of wild-type and mutant p53. FASEB J. 1993, 7, 855–865.
2.Lane, D.; Levine, A. p53 Research: The Past Thirty Years and the Next Thirty Years. Cold Spring Harb. Perspect. Biol. 2010, 2, 000893
3.Shigeto Nishikawa and Tomoo Iwakuma, Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials, Cancers 2023, 15, 429.