Lancet Neurology:Patisiran治疗遗传性甲状腺素运载蛋白介导的多发性神经病性淀粉样变的长期安全性和有效性结果
时间:2022-10-13 15:58:29 热度:37.1℃ 作者:网络
遗传性甲状腺素运载蛋白介导的淀粉样变性,也称为ATTRv淀粉样变性,是一种罕见的遗传性进行性疾病。这种病变由甲状腺素运载蛋白(TTR)基因突变引起,导致TTR错误折叠并在多个器官和组织中(躯体和自主神经以及心脏等)积累为淀粉样沉积物。大多数患者发展为多发性神经病和心肌病的混合表型,并且在临床发病后表现出快速进展。未经治疗的患者的中位生存期为确诊后4.7年,而出现心肌病变的患者的中位生存期为3.4年[95% CI 2.7-5.3]。
这种疾病预后不良的危险因素包括晚期多发性神经病、非Val30Met(也称为p.Val50Met)基因型迟发性疾病(>50岁)和心脏受累。原位肝移植和TTR稳定剂等治疗可能会减缓疾病早期的自然进展,但也经常观察到患者神经功能和生活质量(QOL)的恶化。本研究主要评估Patisiran (一种抑制TTR产生的RNA干扰治疗剂)对遗传性甲状腺素运载蛋白介导的多发性神经病患者长期治疗的安全性和有效性。
这项多中心、开放标签扩展(OLE)试验在19个国家的43家医院或临床中心招募了患者。如果患者完成了APOLLO 3期或OLE 2期研究并且耐受研究药物,则符合条件。来自APOLLO(Patisiran组和安慰剂组)和第2阶段OLE(Patisiran组)研究的符合条件患者参加了这项全球OLE试验。在212名符合条件的患者中,211名患者入选:APOLLO-Patisiran组137名患者、APOLLO-安慰剂组49名患者和OLE-Patisiran2期组25名患者,每3周静脉输注Patisiran0.3 mg/kg或安慰剂,并计划持续5年。其疗效评估包括多发性神经病(改良神经病变损害评分+7 [mNIS+7])、生活质量、自主症状、营养状况、残疾、行走状况、运动功能和心脏压力的测量。全球OLE正在进行中,目前的研究结果是基于对数据截止时已完成12个月疗效评估的患者的中期分析。
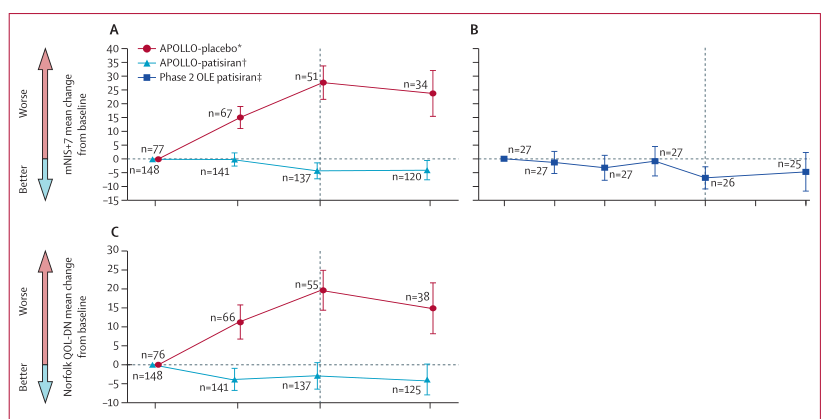
截至2018年9月24日,APOLLO-Patisiran组的137名患者中有126名(92%)完成了12个月的评估,APOLLO-安慰剂组的49名患者中有38名(78%)完成了评估,OLE-Patisiran2期组的25名患者中有25名(100%)完成了评估。在12个月时,患者mNIS+7的改善从研究基线持续到全球OLE治疗(APOLLO-Patisiran组患者平均变化-4.0,95 % CI -7.7至0.3;OLE-Patisiran2期组-4.7,95 % CI -11.9至2.4)。总的来说,211例患者中有204例(97%)报告了不良事件,82例(39%)报告了严重不良事件,23例(11%)死亡。APOLLO-安慰剂组的严重不良事件发生率(49例中28例(57%)高于APOLLO-Patisiran组(137例中48例(35%)或OLE-Patisiran2期组(25例中6例(24%)。
综上所述,全球OLE研究12个月中期数据分析的总体结果表明,Patisiran对遗传性甲状腺素运载蛋白介导的多发性神经病性淀粉样变患者有持续的益处。目前的数据还强调了Patisiran在病程中早期治疗的重要性,以阻止或逆转多发性神经病、自主神经障碍、残疾、营养不良和QOL的进展。
Adams, DavidVita, Giuseppe et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. The Lancet Neurology, Volume 20, Issue 1, 49 - 59

