FDA在2022年第三季度批准了哪些创新药(下)?
时间:2022-10-13 15:52:11 热度:37.1℃ 作者:网络
第一部分见:FDA在2022年第三季度批准了哪些创新药(上)?,以下为第二部分,同样由梅斯小编整理
三、眼科相关
1、FDA已经批准IHEEZO(3%的盐酸氯普鲁卡因眼用凝胶)用于眼表麻醉
9月27日,Harrow与Sintetica共同宣布,FDA已经批准IHEEZO(3%的盐酸氯普鲁卡因眼用凝胶)用于眼表麻醉,成为了近14年来FDA获批上市的首款眼部麻醉剂,也代表着盐酸氯普鲁卡因在美国眼科市场的首次获准使用。
IHEEZO是一种无菌的、不含防腐剂的凝胶滴眼液,无需注射给药。Harrow从Sintetica获得了该药物在美国和加拿大市场的授权。IHEEZO受FDA橙皮书的专利保护,有效期至2038年。
IHEEZO的有效性和安全性在3项临床试验中得到了证明。研究1和2是随机、双盲、安慰剂对照研究,评估了IHEEZO对健康志愿者的影响;研究3是一项随机、前瞻性、多中心、活性对照、观察者掩蔽的III期研究,旨在评估IHEEZO在白内障手术中作为局部麻醉剂的临床疗效。
研究3结果显示,IHEEZO不仅起效迅速(约1-1.5分钟),而且能为手术过程提供足够的麻醉时间(平均持续22分钟)。值得一提的是,使用IHEEZO标准剂量的患者都不需要进行补充治疗。
总体上,IHEEZO的安全性和耐受性良好,最常见的不良反应是瞳孔散大(约25%)。
2、FDA批准Omlonti用于开角型青光眼、高眼压症
2022年9月22日,美国食品和药物管理局(FDA)批准Omlonti®(omidenepag isopropyl眼用溶液,由Santen Inc.公司生产)0.002%用于降低原发性开角型青光眼或高眼压症患者的高眼压(IOP)。
Omlonti是一种相对选择性前列腺素E2(EP2)受体激动剂,旨在通过常规(或小梁)和葡萄膜巩膜外流通道增加房水引流。该批准基于3项随机对照临床试验的数据,这些试验针对的是平均基线眼压为24至26毫米汞柱的开角型青光眼或高眼压症患者。
3、FDA批准CIMERLI与Lucentis互换
CIMERLI是全球首个FDA批准的可互换生物类似药,被批准可与诺华Lucentis(ranibizumab注射剂)互换,适用于Lucentis在美国获批的所有适应症;
四、罕见病相关
1、FDA批准Xenpozyme(olipudase alfa)上市,用于静脉输注治疗酸性鞘磷脂酶缺乏症
8月31日,美国FDA宣布批准赛诺菲(Sanofi)旗下Genzyme公司的创新酶替代疗法Xenpozyme(olipudase alfa)上市,用于静脉输注治疗酸性鞘磷脂酶缺乏症(acid sphingomyelinase deficiency,ASMD)的成人和儿童患者。此药品为FDA批准的首款用于ASMD患者非中枢神经系统症状的药物。
Xenpozyme是一款酶替代疗法,用于替代缺失或有缺陷的酸性鞘磷脂酶。此药物可降解鞘磷脂,进而帮助减少此脂质于肝脏、脾脏与肺脏的累积。Xenpozyme获得美国FDA的快速通道资格、突破性疗法认定、优先审评资格以及孤儿药资格。
Xenpozyme获得美国FDA的批准是基于一项随机、双盲、安慰剂为对照组的试验,共有31位ASMD患者入组。总体而言,此药物可有效改善患者的肺部功能,并减少肝脏与脾脏的大小。Xenpozyme最常见的副作用包含头痛、咳嗽、发烧、关节疼痛、腹泻以及低血压,且有可能造成严重的过敏反应。
2、FDA批准特利加压素(TERLIPRESSIN)用于成人肝肾综合征 (HRS)
9月14日,Mallinckrodt宣布FDA批准Terlivaz(terlipressin,特利加压素)注射液用于治疗成人肝肾综合征(HRS)。这是全球首个获FDA批准用于改善成人肝肾综合征 (HRS) 患者肾功能的疗法。
批准是基于CONFIRM研究,该研究是在2016年7月13日~2019年7月24日期间开展的一项3期、多中心、多国(美国和加拿大)的随机双盲研究。
研究的纳入标准为①肝硬化和存在腹水;②肾功能快速恶化,其定义为血清肌酐水平>2.25mg/dL,并在2周内血肌酐水平翻倍;③停用利尿剂后,血浆容量扩大伴白蛋白,并且48小时内肾功能无改善(血肌酐下降20%或<2.25mg/dL);④若患者条件允许,则在随机分组前应用米多君或奥曲肽;⑤患者年龄≥18岁。
研究的主要排除标准是①血清肌酐水平>7.0mg/dL;②在随机分组后的2天内,进行了至少1次大容量(4L)穿刺;③脓毒症和/或失控的细菌感染;④休克;⑤使用/暴露于肾毒性药物;⑥蛋白尿>500mg/d;⑦血尿或镜下血尿;⑧妊娠;⑨在4周内接受肾脏替代治疗(RRT)等。
纳入研究的患者分为2组,特利加压素组的患者注射特利加压素(0.85mg),对照组注射安慰剂(生理盐水),2组患者每6h接受1次注射。
主要终点:HRS患者肾功能缓解率,其定义为连续2次血肌酐水平≤1.5mg/dL,其间隔时间至少2h,采样时间至少在给药24h后进行。次要终点为①血肌酐达标率:14d或出院后,患者血肌酐水平≤1.5mg/dL;②HRS逆转率:定义为30d后,患者不使用RRT;③全身炎症反应好转率:其定义为14d或出院后,患者全身炎症反应综合征(SIRS)是否好转;④HRS未复发率:15d或出院后证实HRS以出现恢复,且30d时患者未复发。
共计纳入300例患者,其平均年龄(SD)为53.8(11.49),男性占比59.7%。特利加压素组在肾功能缓解率、血肌酐达标率、HRS逆转率、全身炎症反应好转率以及HRS未复发率方面优于对照组(表1)。
表1 CONFIRM的研究结果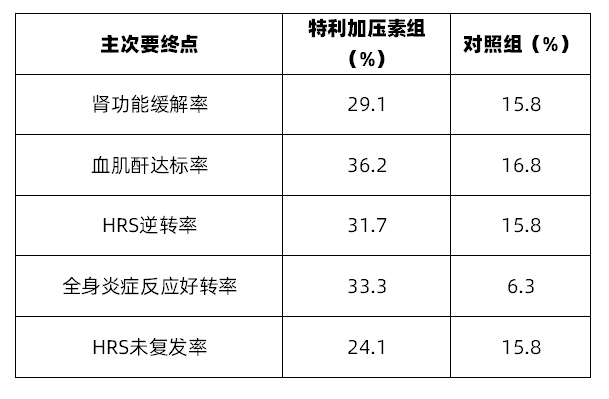
基于该研究结果,特利加压素在美国的药品说明书进行了更新。在更新后的特利加压素说明书中,特利加压素适用于肾功能快速下降的HRS患者,但对于血肌酐水平>5mg/dL的患者,特利加压素带来的获益受限。说明书中建议特利加压素的起始剂量为0.85mg/次,6h注射1次。第1~3d时,每次静注时间应>2min。第4d后,使用剂量应根据血清肌酐水平调整剂量。
3、FDA基因疗法Skysona用于减缓4-17岁早期活动性脑肾上腺脑白质营养不良男孩的神经功能障碍
2022年9月16日, bluebird bio(蓝鸟生物)宣布美国FDA已加速批准其基因疗法Skysona上市,用于减缓4-17岁早期活动性脑肾上腺脑白质营养不良男孩的神经功能障碍的进展。
Skysona(elivaldogene autotemce,eli-cel)是一种一次性给药基因疗法,它通过Lenti-D慢病毒载体(LVV)进行离体转导,将ABCD1基因的功能性拷贝添加到患者自身的造血干细胞(HSC)中。因此患者能够产生ALD蛋白(ALDP),该蛋白有助于分解超长链脂肪酸(VLCFA)。其治疗目标是阻止CALD的进展,并尽可能保留神经功能,包括保留患者的运动功能和沟通能力。该疗法有望终身有效,且无需来自他人的造血干细胞移植。
此次Skysona的批准是基于II/III期临床研究ALD-102及III期临床研究ALD-104的数据支持。两项研究均为开放标签的单臂研究,纳入早期活动性CALD的患者。该研究通过比较Skysona治疗的患者与疾病自然史人群的无主要功能障碍(MFD)生存来进行疗效评估。Skysona治疗的患者在首次NFS≥1的24个月内估计有72%的无MFD生存的可能性,而未经治疗的患者估计无MFD生存的可能性仅为43%。90%的患者在24个月时达到主要评估终点,即无严重功能障碍(Major Functional Disabilities-free)的状态下生存。安全性方面,最常见的非实验室类不良反应包括黏膜炎、呕吐、发热性粒缺、脱发、食欲下降和腹痛等。而实验室类不良反应包括白细胞减少、淋巴细胞减少、血小板减少、中性粒细胞减少、贫血等。
4、FDA批准Zynteglo用于治疗β地中海贫血
8月17日,美国FDA批准Zynteglo用于治疗β地中海贫血。这是第一个针对需要定期输血的 β地中海贫血患者的基因疗法。一次性治疗费用约1984万元。
Zynteglo是一种一次性基因疗法,通过将修饰形式的 β-珠蛋白基因(βA-T87Q-球蛋白基因)的功能拷贝添加到患者自身的造血干细胞中,以生产修饰的功能性成人血红蛋白(HbAT87Q)。一旦患者具有 βA-T87Q-球蛋白基因,他们就有可能将 ZYNTEGLO 衍生的成人血红蛋白 (HbAT87Q) 和总血红蛋白增加到正常或接近正常水平,从而无需定期输血。Zynteglo最早于2019年5月获EMA批准上市。
详细:FDA 批准 Beti-Cel 基因治疗儿科和成人输血依赖型 β-地中海贫血
4、FDA批准Relyvrio用于肌萎缩侧索硬化症患者
2022年9月29日,美国食品药品监督管理局(FDA)批准Amylyx公司Relyvrio(苯丁酸钠和牛磺酸二醇口服固定剂量配方,AMX0035)用于治疗肌萎缩侧索硬化症(ALS)成人患者,商品名为Relyvrio。
该药物是首款在随机、安慰剂对照临床试验中显著延缓ALS疾病进展并能延长生存期的治疗药物,也是116年以来FDA批准的第3款ALS治疗药物。在6个月随机化阶段结束时,接受Relyvrio治疗的ALS患者运动功能下降显著减缓。对所有随机化受试者长达3年的随访显示,与接受安慰剂治疗的患者相比,死亡风险降低了44%。
详细:FDA批准肌萎缩侧索硬化(ALS)复方药物Relyvrio,5年来首个新药
五、神经与精神类药物
1、FDA批准Auvelity(右美沙芬+安非他酮)缓释片用于治疗抑郁症(MDD)成人患者
8月19日,美国FDA已经批准Auvelity(右美沙芬+安非他酮)缓释片上市,用于治疗抑郁症(MDD)成人患者。
Auvelity的疗效和安全性在包含超过1100名抑郁症患者的临床开发项目中得到验证。在名为GEMINI的3期临床试验中,接受Auvelity治疗的抑郁症患者与安慰剂组相比,评定抑郁症状的MADRS量表评分在第6周时获得显著改善。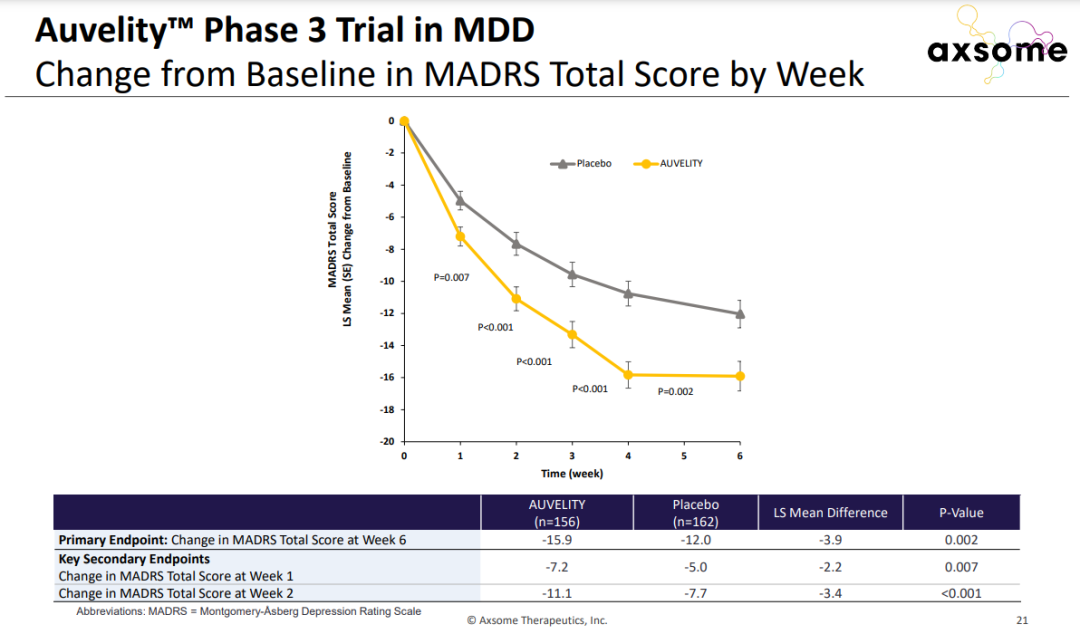 Auvelity在3期临床试验中显著改善患者抑郁症状
Auvelity在3期临床试验中显著改善患者抑郁症状
详细见:FDA批准重度抑郁口服速效药物Auvelity(右美沙芬与安非他酮缓释复方制剂),1周起效!
2、FDA批准SAINT系统来治疗重度抑郁
2022年9月6日,马格努斯医疗(Magnus Medical)宣布,他们研法的SAINT神经调节系统(此前也称为斯坦福神经调节法或SNT)获得了FDA的许可,用于治疗重度抑郁症。
研究人员开展了一项随机双盲实验,参试者为目前正在经历中度至重度抑郁发作的治疗抵抗性抑郁症患者,他们被随机分配为两组,一组接受SAINT治疗(积极治疗组),一组为对照组,主要终点是治疗4周后,患者蒙哥马利-奥斯伯格抑郁评分量表(MADRS)的评分。
试验结果显示,在积极治疗组中79%的人从抑郁症中进入缓解期。相比之下,对照组的这一比例仅为13%。马格努斯医疗在一份新闻稿中说,新的“个体化”神经刺激为治疗抑郁症提供了一种新的选择。
详细见:治疗重度抑郁症的新曙光:美国FDA批准了一种神经刺激疗法SAINT
3、FDA批准SODIUM OXYBATE (γ -羟基丁酸钠)用于治疗成人嗜睡症患者的过度日间嗜睡 (EDS) 或猝倒
2022年7月18日,FDA批准SODIUM OXYBATE (γ -羟基丁酸钠),商品名为LUMRYZ用于治疗成人嗜睡症患者的过度日间嗜睡 (EDS) 或猝倒
六、其它类
1、FDA批准Vibrant治疗便秘
9月3日,FDA批准Vibrant治疗便秘上市,Vibrant由胶囊、激活器和APP组成,胶囊相当于日常复合维生素的大小。
Vibrant使用非常方便,先将胶囊在激活器中激活,然后用一杯水将其吞下(和吃药丸一样)。当胶囊通过肠道时,它会发出低强度的振动来刺激肠神经系统,通过肠-脑轴的信号传导,重新让生物钟同步,恢复自然的排便生物节律。从而改善肠道的蠕动,缓解便秘症状,并提高患者生活质量。
Vibrant在美国完成一项3期临床研究,这项研究在美国90多个临床中心进行,招募312名患者。
试验结果达到了每周完全自发排便(CSBM)的所有主要终点,并且与安慰剂相比,次要终点有显著改善。
与基线相比,接受Vibrant治疗的患者中有39.26%每周有一次或多次额外的每周完全自发排便,有22.7%每周有两次或更多额外的每周完全自发排便,而安慰剂组中的比例仅为22.15%和11.41%。数据显示,与安慰剂相比,次要终点在粪便稠度、生活质量和紧张方面有显著改善。
安全性方面,与安慰剂组相比,Vibrant的不良事件发生率较低(6.5%对9.4%),未报告严重不良事件。
2、FDA批准放射微球Y-90--Eye90 microspheres上市
2022年9月14日,ABK Biomedical宣布旗下的放射微球Y-90--Eye90 microspheres获FDA批准上市,用于治疗动静脉畸形和高血管肿瘤患者。
Eye90 microspheres是第一款具备X射线下显影功能的放射性微球,其解决传统放射性微球巨大弊端,X射线下不可见。让介入医生能够DSA或者CT下实施监测Eye90 microspheres流向,避免发生异位栓塞。
4、FDA批准BLUDIGO(靛蓝磺酸钠)用于泌尿外科和妇科开放性、机器人或内窥镜手术后成人输尿管完整性膀胱镜评估中的可视化辅助工具
2022年7月8日,FDA批准BLUDIGO(靛蓝磺酸钠)用于泌尿外科和妇科开放性、机器人或内窥镜手术后成人输尿管完整性膀胱镜评估中的可视化辅助工具。BLUDIGO的主要活性成分为INDIGOTINDISULFONATE SODIUM。
5、FDA批准辉瑞和莫德纳针对奥密克戎BA.4、BA.5新冠疫苗获美紧急使用授权 (EUA)
美国食品药品监督管理局(FDA)8月31日宣布,批准莫德纳和辉瑞-BioNTech二价新冠疫苗的紧急使用授权 (EUA)。以授权将疫苗的二价制剂用作单次加强剂、或在初次加强疫苗接种后至少两个月后使用。
详细:FDA批准辉瑞和莫德纳针对奥密克戎BA.4、BA.5新冠疫苗获美紧急使用授权 (EUA)
6、FDA 批准伊布替尼用于儿童慢性 GVHD 患者
根据 1/2 期 iMAGINE 试验的结果,FDA 已批准伊布替尼用于患有儿童慢性移植物抗宿主病的患者。
FDA 已批准伊布替尼 (Imbruvica) 作为口服混悬剂或胶囊和片剂形式用于 1 岁或以上患有慢性移植物抗宿主病 (GVHD) 且有 1 个或多个先前失败线经验的儿科患者。
批准的结果基于 1/2 期 iMAGINE 试验(NCT03790332),该试验招募了 1 至 22 岁的中度至重度 GVHD 患者(n = 47)。到第 25 周的总体反应率为 60%(95% CI,44%-74%),中位反应持续时间为 5.3 个月(95% CI,2.8-8.8)。从第一次反应到死亡或慢性 GVHD 的新全身治疗事件,中位时间为 14.8 个月(95% CI,4.6-不可评估)。
总体而言,如果患者有单一器官泌尿生殖系统受累作为慢性 GVHD 的唯一表现,则被排除在外。人群中的中位患者年龄为 13 岁(范围为 1-19 岁),其中 70% 的患者为男性。
对于 12 岁或以上的患者,ibrutinib 的推荐剂量为 420 mg 每天口服一次,对于 12 岁以下的患者,推荐剂量为 240 mg/m2 每天一次。患者可以继续治疗直至疾病进展、复发或出现不可接受的毒性。
参考资料 :
https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=reportsSearch.process

