Genet Med:KDM1A失活导致遗传性食物依赖型库欣综合征
时间:2022-03-09 10:25:15 热度:37.1℃ 作者:网络

背景:原发性双侧大结节性肾上腺增生(PBMAH)是由双侧良性肾上腺皮质肿瘤引起的皮质醇过多(库欣综合征)的肾上腺原因。肿瘤生长缓慢,通常在40岁至60岁之间被诊断。皮质醇过多的临床体征或肾上腺偶发瘤患者可诊断为PBMAH。(3)双侧肾上腺大结节状肾上腺增生(PBMAH)是由双侧良性肾上腺皮质肿瘤引起的皮质醇过多(库欣综合征)的肾上腺原因。肿瘤生长缓慢,通常在40-60岁之间确诊。事实上,大约15%的肾上腺偶发瘤是双侧的,而PBMAH在其中占了相当大的比例。考虑到肾上腺偶发瘤在4%到7%的人群中被观察到,PBMAH的真实发病率可能被低估了。然而,如今诊断的频率要高得多。这种频率的增加导致临床观察到PBMAH是一种不同程度的皮质醇过多和肾上腺增大的异质性疾病,5具有挑战性的诊断和治疗建议。由于肾上腺结节生长缓慢和进行性皮质醇失调,诊断很可能是在发病数年后做出的。手术是双侧或单侧肾上腺切除术最常见的治疗方法。手术适应症基于皮质醇过量水平(考虑其长期临床后果)和肾上腺显像。
在了解PBMAH的遗传学基础方面取得了进展,目的是通过家族性筛查提供早期诊断。事实上,已经确定了PBMAH患者亚群的胚系易感致病变异。最常见的遗传改变是肿瘤抑制基因ARMC5的种系失活变体,在20%到25%的明显散发性PBMAH患者,50%的手术患者和80%有明显家族史的患者中被发现。对ARMC5变体的家族筛查显示疾病的外显率很高。确实,具有ARMC5致病变体的亲属通常存在未诊断的PBMAH。对这些亲属的调查通常会发现皮质醇轻度过多,并伴有长期并发症,甚至是以前没有诊断过的明显的库欣综合征,这是罕见疾病的常见情况。除了ARMC5致病变异外,PBMAH患者可能偶尔会出现导致多肿瘤综合征的基因改变,包括MEN1、APC和FH基因。然而,大多数明显散发性PBMAH患者在这些基因中没有出现任何致病变异。
目的:探讨原发性双侧大结节性肾上腺增生(PBMAH)患者出现食物依赖型库欣综合征(FDCS)的遗传原因,并探讨肾上腺葡萄糖依赖性胰岛素样多肽受体的异位表达。据报道,大约25%的PBMAH指数病例中有种系ARMC5改变,但在FDCS患者中没有。
方法:对36例肾上腺切除患者的PBMAH组织进行多组学分析(RNA测序、单核苷酸变异体阵列、甲基组、miRNome、外显子组测序)。
结果:综合分析发现3个具有不同临床特征的分子群,即G1(16例ARMC5失活变异型)、G2(6例葡萄糖依赖性促胰岛素多肽受体异位表达的FDCS)和G3(14例表型较轻的患者)。外显子测序发现5例G2患者KDM1A基因发生胚系截短变异,并持续存在1p上KDM1A野生型等位基因的体细胞缺失,导致KDM1A在信使RNA和蛋白水平的表达均丢失(P分别为1.2×10-12和P<0.01)。随后,在4个附加的FDCS指标病例中有4个发现了KDM1A致病变异。

图1原发性双侧大结节性肾上腺增生(PBMAH)肿瘤的转录组和染色体改变。答:根据转录组图谱对PBMAH肿瘤进行的非监督聚类发现了3个表达簇:E1簇(蓝色)包含所有具有ARMC5致病变异的肿瘤,E2簇(红色)包括术前诊断为典型的食物依赖型库欣综合征(FDCS)患者的所有肿瘤,E3簇(绿色)包含来自没有种系ARMC5致病变体或FDCS的患者的肿瘤(补充表1A和2)。中间的面板显示了各组之间表达差异最大的信使RNA的热图(补充表3)。底部显示了GIPR和KDM1A在每个肿瘤中的表达。另见补充图3.B。PBMAH肿瘤的染色体改变。染色体得失的累积比例由染色体定位(TOP)提供。每个患者的染色体增加和丢失都显示出来(下图)(补充表2和6)。损失用蓝色表示,收益用红色表示,cnLOH用紫色表示。CnLOH,拷贝中性杂合性丢失。
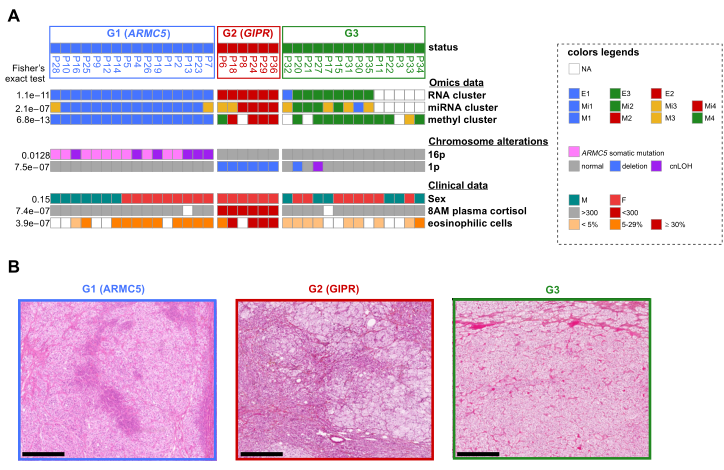
图2多重组学、临床和病理数据确定了3组患者。答:提供了组学分组之间的联系(补充表2);最常见的染色体改变(16p和1p,补充表2和6)、病理特征和临床注释(补充表1A)(使用费舍尔精确检验来确定P值)。G1组(蓝色)包含来自ARMC5胚系致病变种患者的肿瘤,其特征为更严重的库欣综合征和更大的肾上腺,如表1所示,具有一致的体细胞16p cnLOH(三分之一的肿瘤)或体细胞ARMC5失活变体;G2组(红色)包含来自6名GIPR过表达的患者的肿瘤,1p的体细胞杂合性持续丧失,由于FDCS,早晨血浆皮质醇显著低于其他两组;显微图像显示三组之间的形态差异。1例ARMC5致病变异型患者肿瘤嗜酸性细胞<20%(左),1例GIPR过表达患者肿瘤嗜酸性细胞>30%(中),1例第3组患者肿瘤嗜酸性细胞<5%。H&E藏红花。标尺=500m.cnLOH,拷贝中性杂合性丢失;F,雌性;H&E,苏木素和曙红;M,雄性;miRNA,microRNA;NA,不可用。

图3 KDM1A致病变异体和KDM1A免疫组织化学。A.在患者中发现的各种基因改变的KDM1A蛋白和互补DNA(NM_015013)中的位置。在最初的队列中发现的致病变异用红星表示,在另外4个指征病例中识别的变异用紫星表示。黄色三角形代表NM_001009999亚型中2个补充短外显子的位置(分别为60和12个碱基对)。这两个外显子在本研究中均未发生改变。外显子15的Sanger测序获得的电泳图谱显示,在患者P36中发现的变异在白细胞DNA中处于杂合状态,在肿瘤DNA中处于继发于野生型1p等位基因丢失的半合子状态。原发性双侧大结节肾上腺增生症患者中有1例出现KDM1A免疫组织化学染色。在GIPR组(G2)的4例患者中,大多数细胞核KDM1A在肿瘤中的染色比邻近的非肿瘤肾上腺弱或没有。而ARMC5组(G1,n=4)和G3组(G3,n=4)的肿瘤细胞均有KDM1A染色。KDM1A免疫组织化学染色。比例尺=50μm。
结论:KDM1A失活可解释约90%的FDCS PBMAH。现在可以为大多数PBMAH手术患者及其家属提供ARMC5和KDM1A的基因筛查,为早期诊断和改善管理开辟了道路。
原文出处:Vaczlavik A, Bouys L, Violon F,et al.KDM1A inactivation causes hereditary food-dependent Cushing syndrome.Genet Med 2022 Feb;24(2)

