【综述】| TCR-T免疫治疗肿瘤:现状、挑战及展望
时间:2023-09-06 10:53:38 热度:37.1℃ 作者:网络
[摘要] T细胞受体工程T细胞(engineered T cell receptor-T cell,TCR-T)疗法和嵌合抗原受体T细胞(chimeric antigens receptor-T cell,CAR-T)疗法是目前过继性T细胞治疗最有效的两种方式。由于CAR仅能识别肿瘤表面的抗原,在实体瘤治疗中至今未有令人满意的结果。TCR不仅能识别肿瘤表面抗原,同时能识别胞内抗原,因此,TCR-T疗法在治疗实体瘤方面显示出前所未有的前景,成为极具潜力的治疗方式。本综述探讨了TCR-T疗法与CAR-T疗法识别癌症抗原机制的差异及当前TCR-T疗法靶向的临床靶点和不同类型的肿瘤抗原,描述了TCR-T抗肿瘤治疗的临床开发现状,并讨论了临床前评估TCR效价的标准和目前TCR-T治疗的优势、存在的局限性及可能有效的应对措施。最后,我们回顾了TCR-T治疗的现状和当前仍存在的一些挑战,强调靶向肿瘤特异性抗原的重要性,概述了结合检查点阻断治疗和溶瘤病毒等的新抗原特异性TCR-T治疗策略,以期这种联合治疗能够显著改善癌症的免疫治疗效果,并对未来TCR-T治疗根除多发性癌症提供一些思路。
[关键词] T细胞受体工程T细胞;免疫治疗;实体瘤;肿瘤抗原;嵌合抗原受体T细胞
[Abstract] Engineered T cell receptor-T cell (TCR-T) therapy and chimeric antigen receptor-T cell (CAR-T) therapy are currently the two most effective ways of adoptive T cell therapy. Because CAR can only recognize antigens on the surface of tumors, CAR-T therapy has not yet had satisfactory results in the treatment of solid tumors. TCR can not only recognize tumor surface antigens, but also intracellular antigens. Thus TCR-T therapy has shown unprecedented promise in the treatment of solid tumors, and has become an extremely attractive treatment modality. This review described the differences between TCR-T therapy and CAR-T therapy in recognizing cancer antigens, the clinical targets and different types of tumor antigens targeted by current TCR-T therapy, the clinical development status of TCR-T antitumor therapy, and discussed the criteria for preclinical evaluation of TCR titer and the advantages, limitations and possible effective countermeasures of current TCR-T therapy. Finally, we reviewed the current status of TCR-T therapy and some of the challenges, emphasized the importance of targeting tumor-specific antigens, and outlined neoantigen-specific TCR-T treatment strategies combining checkpoint blockage therapy and oncolytic viruses, which we expect will significantly improve cancer immunotherapy and provide some clues for future TCR-T therapy to eradicate multiple types of cancer.
[Key words] Engineered T cell receptor-T cell; Immunotherapy; Solid tumors; Tumor antigens; Chimeric antigens receptor-T cell
过继性T细胞治疗(adoptive T cell therapy,ACT)策略已成为癌症治疗的主要方式。T细胞受体工程T细胞(engineered T cell receptor-T cell,TCR-T)疗法和嵌合抗原受体T细胞(chimeric antigens receptor-T cell,CAR-T)疗法是将T细胞特异性重定向到肿瘤抗原的两种主要方法[1],作为最新、最有效的两种免疫治疗技术,近年来受到了广泛关注。在临床试验中,虽然CAR-T疗法在B细胞恶性肿瘤治疗方面疗效显著[2],但对实体瘤的治疗却未能达到令人满意的效果[3],而TCR-T疗法更适用于实体瘤的治疗[4],主要是由于两者识别抗原的机制存在差异。首先,两者识别位置不同,CAR-T的识别仅限于表面抗原,而由于跨膜蛋白约仅占蛋白质组的1/4,因此,可靶向CAR-T疗法的抗原相当有限,且CAR-T激活需要更高浓度的靶抗原[5]。TCR-T不仅能识别细胞膜抗原,还能识别肽-主要组织相容性复合体(peptide-major histocompatibility complex,pMHC)呈递的细胞内肿瘤抗原[6],不受靶细胞表面抗原表达的限制,从而能更广泛地识别靶抗原,诱导更持久的免疫突触形成过程,TCR还可被单一的靶抗原分子激活,对低拷贝数抗原比CAR-T更敏感。此外,两者识别方式不同,CAR通过抗体的单链可变片段识别抗原,TCR通过由α和β肽链组成的异二聚体来识别pMHC分子呈递的抗原(图1),TCR-T识别肿瘤细胞后被激活,其分泌细胞因子、端粒酶、穿孔素杀伤肿瘤细胞。而多项临床试验[7-8]也证明TCR-T疗法在血液系统肿瘤和实体瘤中安全性和有效性良好。1988年,Blüthmann等[9]首先将TCR基因从一个T细胞转移到另一个T细胞中,使第二个T细胞具有相同的抗原特异性,拉开了TCR基因治疗的帷幕。到目前为止,有关TCR-T对于血液病及实体瘤治疗的研究呈逐年上升趋势。
本综述将详细描述TCR-T治疗的基本机制和其靶向的不同类型的肿瘤抗原,讨论临床前评估TCR的依据和临床开发现状,以及TCR-T在实体瘤治疗中的优势、存在的挑战及应对措施,并对TCR-T治疗进行展望。
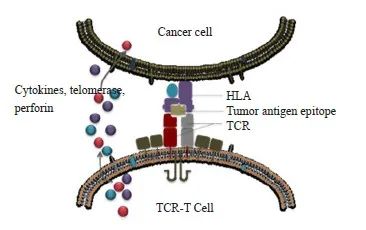
图1 TCR-T识别肿瘤细胞后被激活,其分泌细胞因子、端粒酶、穿孔素杀伤肿瘤细胞
Fig. 1 Having recognized tumor cells, TCR-T is activated and they secret cytokines, telomerase and perforin to attack and kill tumor cells
1 TCR靶向的肿瘤抗原及其特点
选择理想的抗原是提高抗肿瘤效率和降低相关毒性的关键,肿瘤特异性、免疫原性是选择抗原的首要考虑因素。通常,人类肿瘤抗原主要分为肿瘤相关抗原(tumor-associated antigen,TAA)和肿瘤特异性抗原(tumor-specific antigen,TSA)两大类[10]。表面抗原通常是TAA,正常组织也可以表达这些抗原来影响功能,包括癌-睾丸抗原、过表达抗原和分化抗原。而近90%的实体瘤靶向依赖于TSA,其可分为新抗原和癌病毒抗原。尽管TCR-T可以靶向所有肿瘤抗原,但肿瘤抗原的多样性往往会引起“靶上,肿瘤外”毒性[11]。迄今为止已确定的有足够安全性和有效性的靶点数量仍然有限,通常选择在肿瘤中高表达但在正常组织中低表达的靶抗原来限制潜在的脱靶效应[12]。
1.1 TAA
TAA是由癌细胞表达的自身抗原,在肿瘤细胞和健康组织细胞中均有表达。因此,研究针对TAA的疗法应结合T细胞介导的潜在靶向脱瘤毒性。黑色素瘤分化抗原是迄今为止发现较早的肿瘤抗原之一,且分化抗原通常形成“靶上,肿瘤外”毒性的风险最大。例如,针对由T细胞识别的黑色素瘤相关抗原-1(melanoma-associated antigen-1 recognized by T cells,MART-1)TCR-T治疗的实验[13]中,靶向DMF5表位的TCR-T研究报告了对耳朵、眼睛及皮肤的毒性反应。而在癌细胞中高表达,在健康细胞上几乎不表达的过表达抗原同样有潜在产生“靶上,肿瘤外”毒性的能力。Wilms肿瘤1(Wilms’ tumor 1,WT1)抗原是被广泛研究的一种抗原,高表达于多种类型的白血病中,靶向WT1抗原的T细胞能够靶向肿瘤细胞,而对正常组织无免疫毒性,有研究[8]证实,过表达抗原在肿瘤和正常细胞间存在的表达水平差异可能足以使T细胞破坏高抗原表达的癌细胞,而对低表达的健康组织的毒性达到最小。目前,WT1抗原也成为TCR-T治疗的理想靶点,靶向WT1抗原的研究已进入临床试验阶段[14]。癌-睾丸抗原如黑色素瘤相关抗原(melanoma-associated antigen,MAGE)-A1、纽约食管鳞状细胞癌-1(New York esophageal squamous cell carcinoma-1,NY-ESO-1)等,由于在许多肿瘤中高表达,在正常组织中很少表达,因此被认为是高效而安全的免疫治疗靶点。Robbins等[15]的一项研究针对NY-ESO-1/人类白细胞抗原(human leukocyte antigen,HLA)-A2的TCR在转移性黑色素瘤和滑膜肉瘤患者中显示出显著的抗肿瘤反应,且没有观察到不良反应,但Morgan等[16]研究显示,靶向MAGE-A3的TCR与在大脑中表达的相关多肽MAGE-A12存在严重的交叉反应。此外,在Cameron等[17]进行的一项试验中,靶向MAGE-A3/HLA-A1的TCR识别与Titin相似但不完全相同的多肽,致使1例黑色素瘤患者和1例骨髓瘤患者出现心血管毒性并死亡。以上研究中TCR与正常组织之间的相互作用显示了靶向癌-睾丸抗原潜在的毒性。
1.2 TSA:新抗原
某些肿瘤特异性突变会产生新的抗原,近年来新抗原作为一类免疫原性TSA被发现[18]。事实上,靶抗原在正常组织或细胞中的表达会导致T细胞对健康组织的破坏,即“靶上,肿瘤外”毒性[19]。而由于新抗原仅在肿瘤细胞中特异性表达,靶向此类抗原可以降低“靶上,肿瘤外”毒性的潜在风险,因此成为TCR-T治疗有吸引力的靶点。大多数随机性的突变并不能在患者之间共享,不能成为抗原靶标。而能促进癌症进展的突变能在特定癌症类型的患者之间共享。KRAS是人类癌症中常见的突变原癌基因之一,KRAS G12D/G12V共同在60%~70%的胰腺癌和20%~30%的结直肠癌中被发现[20]。目前,有关KRAS作为新抗原的研究被大量报道,有研究[21- 22]显示,从KRAS G12V和G12D突变中分离得到的高亲和力TCR转导外周血淋巴细胞,获得的TCR-T可以特异性地靶向肿瘤细胞,并能抑制异种移植模型中的肿瘤细胞生长。有研究[23]报告了1例转移性结直肠癌患者在转移靶向突变KRAS G12D的细胞毒性T淋巴细胞后肿瘤消退。上述研究结果提示KRAS突变抗原特异性TCR是一种值得期待的治疗KRAS突变实体瘤的手段。然而,并不是所有的肿瘤突变都可能导致新的免疫原性抗原。
1.3 TSA:癌病毒抗原
研究[24-25]发现,大多数病毒驱动的癌症是由驱动细胞转化和癌症进展的病毒癌基因的表达所介导。与其他类型的抗原不同,病毒癌基因通常均匀表达于病毒驱动的癌细胞中,而在正常细胞中几乎不表达。因此,癌病毒抗原也成为有前景的肿瘤抗原靶点[26-27]。人乳头状瘤病毒(human papillomavirus,HPV)-16 E7特异性TCR-T在体内外可特异性识别、杀伤HLA-A*0201阳性HPV-16+肿瘤细胞系,且不与人自身多肽发生交叉反应[25],表明癌病毒抗原HPV-16 E7特异性TCR对宫颈癌具有显著作用。目前,癌病毒抗原已被证明是高效有力的抗瘤靶点,众多靶点处于临床研究阶段。
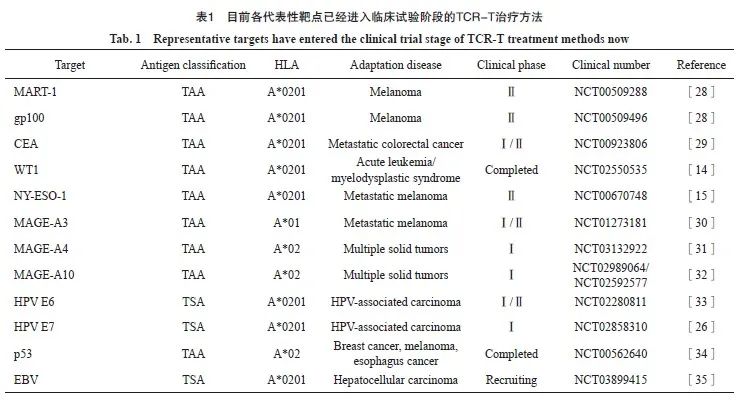
综上可知,抗原的选择是过继T细胞治疗首先应考虑的因素,选择肿瘤特异性、致癌性和免疫原性的理想抗原对增强抗肿瘤效果和降低毒性至关重要。基于每种肿瘤抗原都有不同的适应证及自身特点,目前靶向不同抗原的TCR-T治疗也被大量研究,许多已进入临床试验阶段(表1)。
2 候选TCR的评估标准
在选择进入临床的TCR时,通常会评估以下几个方面。
2.1 亲和力
TCR与MHC之间的高效相互作用是TCR-T成功靶向肿瘤的前提。TCR与MHC之间的结合或TCR-T对靶抗原的特异性识别能力通常用TCR亲和力来表征。使用比DMF4受体亲和力更高的DMF5 TCR,转导后的T细胞在试验中观察到有更好的应答率[30],暗示能够识别肿瘤细胞表面低表达的pMHC的高亲和力TCR被作为更好的选择可用于大多数临床试验。理论上,为增强免疫反应应寻找与靶抗原具有高亲和力的TCR,但自身免疫性疾病通常与高亲和力TCR的治疗相关,因此筛选具有最佳亲和力的TCR极具挑战性。太低的亲和力可能对靶向非肿瘤组织存在潜在的毒性风险,而太高的亲和力可能导致异常的免疫激活,引发细胞因子风暴的产生。而具有中到低等亲和力的TCR可以介导肿瘤的杀伤,且不会引起自身免疫性疾病。确定抗原识别受体的理想亲和力应控制在合适的阈值范围内,已有研究[13]统计T细胞亲和力阈值在5~10 μmol/L时,T细胞活性最大,亲和力大于正常范围(1~100 μmol/L)的TCR更有可能发生交叉反应[13,36]。因此,在进行TCR亲和力优化的同时应合理、准确评估,以免过高亲和力的TCR在临床使用时对患者产生不利影响。
2.2 安全性
尽管TCR-T在治疗实体瘤方面有极大潜力,但其安全性问题同样备受关注。在靶[29]和脱靶[30]肿瘤毒性反应在之前的TCR临床试验中已被报道。上述有关各种靶抗原表达特点的描述,暗示TCR激活的T细胞可能不能正确区分肿瘤细胞和表达靶抗原或类似靶抗原的正常细胞。因此,TCR-T可能会对正常组织产生损害。在靶毒性在TCR识别MAGE-A3和MAGE-A12共有表位的T细胞后发生的致命神经毒性中得到明确[16]。此外,Sanderson等[37]报道了TCR潜在的同种异体反应,发现限制性MAGE-A4 TCR与HLA-A*0205产生了同种异体反应,因此,在候选TCR靶向新的抗原表位进入临床试验前,必须评估其在健康组织内的表达模式和HLA的 递呈。另外,错配的TCR会导致潜在的危险脱靶毒性,TCR错配可能会产生新的TCR,这些TCR没有经过人体胸腺选择,可能具有自身抗原特异性。若机体健康组织与肿瘤细胞有相同或类似的抗原时,错配后的TCR便会产生靶向和肿瘤外毒性。因此,在临床前研究中应该对每种肿瘤抗原进行严格筛查,以确定其在人体组织中的表达程度。
3 TCR抗肿瘤治疗的临床开发现状
2004年,Khong等[38]从黑色素瘤中分离得到第一个TCR,其靶向分化抗原gp100,为癌症治疗提供了新选择,该实验的成功开启了TCR-T治疗时代。自2015年以来,治疗各种实体瘤和血液系统恶性肿瘤的TCR-T研究大量增加,尤其是2017—2019年,TCR-T疗法得到迅速发展,临床试验数量也随TCR-T疗法的需求逐渐增加。虽然基于TCR-肽/MHC相互作用的TCR-T治疗是最常见且应用最广泛的免疫治疗方法,但其他基于TCR的免疫治疗策略也已被探索用于临床试验中,利用一端可溶性TCR和另一端的免疫细胞激活基序所组成的融合蛋白近年来也被研究,2022年1月25日,美国食品药品管理局(Food and Drug Administration,FDA)批准Kimmtrak(tebentafusp-tebn,IMCgp100)用于HLA-A*0201阳性的无法切除或转移性葡萄膜黑色素瘤成人患者的治疗[39],自此第1个获得美国FDA批准的TCR-T治疗产品诞生,其是首次获批治疗实体瘤的双特异性T细胞接合器,也是目前唯一获批治疗不可切除性或转移性葡萄膜黑色素瘤的疗法。Kimmtrak的获批具有里程碑意义,彰显了TCR-T疗法强大的抗肿瘤能力。世界多家机构也积极开展TCR-T免疫治疗的研究,一直努力将各自的研究推向临床治疗阶段。截至2022年10月18日,全球共有710个有关“TCR”的关键词在美国国立卫生研究院的临床试验数据库(www.clinicaltrials.gov)中被检索到,所有检索到的研究大多数针对实体瘤,其中最常见的实体瘤是黑色素瘤,其次是肠癌、胃癌、肺癌等。依据研究靶点分类,主要有包括NY-ESO-1、MAGE家族蛋白、MART-1、癌胚抗原(carcinoembryonic antigen,CEA)等TAA靶点,新抗原包括乙型肝炎病毒(hepatitis B virus, HBV)/HPV、KRAS等。27个临床试验中,就靶抗原分类而言,TAA最多,其次是病毒抗原,新抗原的临床试验也有逐年增加的趋势(表1)。根据靶点的统计,NY-ESO-1、MAGE-A4是临床试验靶向最多的靶点,其次是HPV、MART等。这些临床试验中,最常见的限制性等位基因依次为HLA-A*02、HLA-A*11、HLA-A*24,此外,靶向癌症类型最多的是黑色素瘤。从试验设计和策略的角度来看,大多数临床试验进度是Ⅰ或Ⅱ 期,目前还没有产品达到Ⅲ期。
4 TCR-T在实体瘤治疗中的优势、局限性、面临的挑战及应对策略
4.1 治疗实体瘤的优势
与CAR-T仅识别表面抗原不同,MHC分子呈递的任何抗原均能被TCR-T识别,不管是胞内或胞外抗原,亦或是肿瘤细胞突变后产生的新抗原。因此TCR比CAR能识别更多的靶抗原和肿瘤类型。此外,CAR-T疗法虽不受MHC的限制,但需要在体外大量制备抗原特异性T细胞。CAR-T疗法为正常的T细胞配备了肿瘤细胞表面抗原特异性单链抗体,该单链抗体可连接到CD3ζ的胞内区域,在治疗中使用的抗体的亲和力一般高于TCR,因此,CAR-T的激活需要更高浓度的靶抗原[40],而TCR-T通过基因工程改造,对肿瘤细胞的亲和力增强,可被微量靶抗原激活,故TCR比CAR对低浓度的靶抗原更敏感(图2)。由于实体瘤细胞缺乏细胞表面生物特异性标志物,因此TCR-T比CAR-T更适宜于实体瘤的治疗。此外,TCR-T治疗在试验中观察到比CAR-T介导更少的细胞因子释放[6],因此TCR-T治疗的综合风险评分(comprehensive risk score,CRS)比CAR-T更低。因此,TCR-T在治疗实体瘤方面具有极大潜力,近年来TCR-T疗法的一些临床试验被大量研究。目前,Kimmtrak的获批进一步证实了TCR-T疗法在实体瘤中的有效性和安全性。近年来,新抗原作为一类免疫原性TSA被发现,其来自于自身致癌病毒蛋白的肿瘤特异性突变。针对新抗原和病毒蛋白的T细胞不会经胸腺选择,因此很有可能分离出针对这些靶点的T细胞克隆。而此类抗原多表达于细胞内,因此,TCR比CAR更有可能成功地靶向此类抗原。
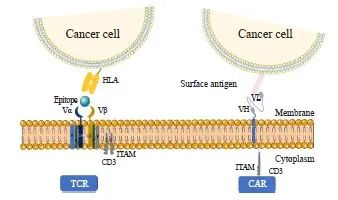
图2 TCR和CAR与肿瘤细胞的识别及作用模式
Fig. 2 The recognition/action mode between TCR and cancer cell and between CAR and cancer cell
4.2 局限性及应对策略
TCR依赖MHC分子的呈递来识别和激活T细胞,TCR-T仅能识别MHC分子呈递的抗原。因此,TCR-T治疗受到MHC分类的限制,任何给定的TCR都只能用于治疗具有相同MHC表达的患者。此外,外源链与内源链之间潜在的错配风险,可能会对机体造成损伤。所以,尽管TCR-T治疗显示出良好的抗瘤潜力,但其在实际临床应用中仍存在诸多局限性。
4.2.1 HLA限制性
HLA编码基因是人类基因组中最具多态性的基因,迄今为止已鉴定出超过20 000个HLA Ⅰ 类等位基因[41]。由于CD8+ T细胞的杀伤能力依赖于TCR对HLA Ⅰ类分子呈递的肿瘤抗原的特异性识别,CD4+ T细胞的调节功能依赖于TCR对HLA Ⅱ类分子呈递的抗原识别,所以TCR的功能只适应于与HLA相匹配的细胞或患者。因此,选择接受TCR-T疗法的患者,不仅必须表达靶向抗原,还必须表达相应的抗原限制性HLA等位基因。然而,TCR-T疗法的使用受限于相对常见的HLA等位基因的TCR。据统计,高加索人在HLA-A*0201中占比最大,中国汉族人在HLA-A*1101中占比最大[42]。因此,目标人群中HLA亚型的流行是TCR产品开发过程中的一个重要考虑因素。对于每种抗原,一个可行的策略是为多个显性不同HLA亚型呈递的表位选择TCR,以覆盖大多数表达该抗原的患者。
4.2.2 TCR-T归巢受限
细胞毒性T淋巴细胞(cytotoxic T lymphocyte,CTL)在其表达的趋化因子受体和肿瘤分泌的趋化因子的相互作用下迁移浸润至实体瘤才能将其根除。当CTL表达的趋化因子受体与肿瘤分泌的趋化因子不匹配时,CTL的归巢过程将会受限,从而影响抗肿瘤效应。研究[43]发现,将CC族趋化因子受体2(CC chemokine receptor 2,CCR2)转导到SV40的抗原特异性TCR-T中,可促进表达CC族趋化因子配体2(CC chemokine ligand 2,CCL2)的转移性前列腺癌细胞的增殖,提高TCR-T的体内抗肿瘤效果。将CXC族趋化因子受体2(CXC chemokine receptor 2,CXCR2)导入MAGE-A3的特异性TCR-T,CXCR2- TCR-T在体内的归巢增强,肿瘤侵袭增强,并优先聚集在肿瘤部位;与对照组相比,CXCR2- TCR-T的存活和肿瘤消退显著增强[44]。Hu等[45]研究发现,化疗也能导致过继转移的T细胞更多地归巢和浸润。除设计带有趋化因子受体的T细胞外,趋化因子可以直接引入肿瘤,增强T细胞抗肿瘤效应[46]。
4.2.3 T细胞在体内的持久性
过继性T细胞在体内的持久性是消除肿瘤、防止复发的关键因素。有临床试验[47]发现,大多数无应答的患者体内回输的特异性T细胞没有表现出应有的持久性,从而降低了疗效。目前,用细胞因子或药物刺激T细胞及对T细胞信号进行遗传修饰等多种方法被开发以增强T细胞的持久性。其中,临床上最常见的方法是在回输T细胞之前进行“清淋”,减少内源性T细胞对稳态细胞因子的消耗[48]。此外,内源性免疫细胞可以充当细胞因子接收器,在ACT使用前将淋巴去除,可以为转移的T细胞节省有限的细胞因子,同时可以消除免疫抑制的调节T细胞(regulatory T cell,Treg)和髓源性抑制性细胞(myeloid-derived suppressor cell,MDSC),提高ACT的持久性和有效性[49]。另有研究[50]表明,重组白细胞介素(interleukin,IL)-2的纳米颗粒递送实现了持久的肿瘤控制,并且在癌症免疫治疗中具有较少的全身性不良反应。此外,通过将激活T细胞信号转导的部分(CD28或4-1BB)的细胞内结构域插入CD3ζ而不是改变TCR亲和力。研究[51]表明,经过修饰的TCR-T具有更好的疗效,可促进体内T细胞的增殖和持久性增强。
4.2.4 免疫抑制肿瘤微环境
实体瘤微环境中的免疫抑制细胞如Treg、MDSC等,分泌IL-10及转化生长因子-β(transforming growth factor-β,TGF-β)等细胞因子,具有诱导恶性肿瘤生长、迁移、逃避免疫系统和T细胞杀伤的作用,因此TCR-T很难彻底清除肿瘤细胞。目前,研究最多的方法是通过将抑制性信号转换为刺激性信号来改变免疫抑制环境对T细胞的抑制作用。Roth等[52]报道NY-ESO-1特异性TCR在转导TGF-β R2-41BB存在的条件下,在体内实体瘤模型中抗肿瘤效应显著增强。此外还有免疫检查点抑制剂合用[53]、MDSC基因掺入[54]等方法。研究[55]表明,肿瘤微环境中的基质细胞能表达免疫检查点,从而抑制T细胞发挥抗肿瘤功能。在一项体外和异种移植模型中,具有CD3和CD28结构域的嵌合开关受体(chimeric switch receptor,CSR)可增强细胞毒作用及细胞因子的产生,促进CAR-T浸润至肿瘤,并靶向MDSC,且没有引起严重的细胞因子风暴。此外,T细胞可以被修饰以产生趋化因子作为有效载荷,促进它们的激活,渗透至肿瘤微环境中。以上研究证实在TCR-T抗肿瘤应用中使用CSR能将肿瘤微环境的各种免疫抑制信号转化为免疫刺激信号。
5 TCR-T免疫治疗面临的挑战及展望
免疫疗法现已成为癌症治疗的主要手段。肿瘤抗原特异性多肽/MHC作为癌症免疫治疗的靶点也已被广泛研究。如今,TCR-T疗法的推广和使用已证明其是治疗癌症的一种高效、安全的免疫疗法。与CAR-T相比,TCR-T具有许多优势,其可识别来自表面和细胞内的蛋白,能够检测到更多的靶点,从而治疗更多的疾病。此外,TCR可被单分子刺激激活,灵敏度高于CAR。多项TCR-T的临床研究[56-58]表明,TCR-T在治疗实体瘤方面具有显著的优越性。从2015年至今,TCR-T治疗的临床试验急剧增长,尤其是Kimmtrak获得美国FDA的批准,进一步表明TCR-T疗法的巨大潜力。尽管众多研究表明TCR-T疗法已成为治疗各种类型癌症的一种有前途的策略,但TCR-T疗法在实际临床中仍存在一些挑战:
⑴ 新抗原在TCR介导的ACT的临床开发中受到了其内在特性的阻碍:① 新的表位是否可以由MHC呈现并被新抗原特异性TCR有效识别。由于新抗原在肿瘤内由某些基因特异性突变产生,但并不是所有的肿瘤突变都可能导致新的免疫原性抗原。Ye等的研究[59]主要在单细胞水平上采用免疫系统的高通量测序(high-throughput sequencing of the immune repertoire,HTS-IR)技术,包括用于重建TCR和识别免疫原性新抗原的TraCeR和单细胞TCRseq等。② 新抗原可被免疫系统特异靶向恶性肿瘤的相关证据令人信服,然而二代测序的准确性和特异性等仅使一小部分新抗原被发现用于癌症的治疗,且在低突变负荷的癌细胞中很难检测到新抗原。对于这一问题, Chen等[60]设计了一种筛查癌症患者新抗原的方法,最终通过新抗原肽库鉴定出了免疫原性新抗原。Joglekar等[61]设计出一种新的抗原发现报告系统,通过将pMHC与CD3ζ-CD28的胞内信号连接进行抗原表位的筛选。综上,随着全外显子组测序、共享新抗原库和巨噬细胞增多症筛查平台等技术的发展,相信未来会有更多的新抗原被发现和确定。
⑵ 使用mRNA或随机整合病毒来传递外源TCR,可能潜在地导致引入的TCR与内源性TCR的错配。TCR错配能导致引入的TCR的表达减少,此外,TCR错配会产生新的未经过胸腺选择的TCR,可能会导致自身抗原特异性。现有以下策略可促进TCRα/β链的正确配对,防止错配的发生:
① 鼠源C区改造:研究[62]发现,小鼠序列替换人的TCR恒定区是减少错配的一种方法,与人类来源的TCR相比,当使用鼠源的TCR时,TCR-T显示出更高的生物活性;② 加入额外的二硫键:通过在C区α残基48位与C区β残基57位再引入一个稳定的二硫键,能够增强TCR的正确配对[63];③ 单链TCR:通过将Vα和Vβ结构域与ScTCR共价连接起来形成单链TCR,通过空间位阻抑制错配也是减少错配的一种方法[64];④ 将人TCR恒定结构域C端α和C端β交换,可使TCR减少错配[65];⑤ 通过基因工程敲除内源性TCR,减少内源性TCR对CD3结合的竞争也能减少TCR错配[66];⑥ 通过CRISPR-Cas9基因编辑,敲除内源性TCRα和β链,并转导单链特异性TCR,也可防止错配[67]。
⑶ 几种有望彻底根除实体瘤的新策略:
① 加强基因工程T细胞向实体瘤的浸润和迁移:研究[68]发现,T细胞具有与肿瘤微环境分泌的趋化因子相匹配的趋化因子受体,有助于T细胞向肿瘤的迁移。因此,可将趋化因子与细胞因子结合[32],通过加入溶瘤病毒[69]或疫苗佐剂来增强TCR-T的抗肿瘤效果。
② 增强对肿瘤的免疫监视作用:回输的TCR-T在体内的存活时间有限,一旦免疫细胞减少,肿瘤细胞就容易发生免疫逃逸导致癌症复发。为此,可通过细胞因子的使用保持T细胞在体内的存活和功能发挥,增强其持久性[70],但此方法无靶向性,易产生不良反应[71]。研究表明,通过TCR-T靶向输送细胞因子可以保持T细胞存活,并能产生记忆性T细胞[29],同时也能恢复肿瘤微环境造成的炎症环境[72],保持T细胞抗肿瘤适宜的环境。
③ 增强结构亲和力:配对和密码子优化能增强蛋白质的表达,可能会增强特异性反应。研究[73]发现,对TCRα和β链的CD3区域引入选择性修饰有助于TCR-T对抗原的识别和结合。另外,减少TCR的糖基化可提高其功能和亲和力,阻止转导的TCR内化。将TCRα链的3个跨膜残基修饰为疏水氨基酸,也能增强TCR的稳定性和表达水平[74]。此外,应用P2A或IRSE元件连接TCRα和β链能提高TCR的表达水平,并能降低诱导自身免疫性疾病的风险[8]。
综上,尽管TCR-T已成为攻克实体瘤有效的免疫治疗手段,但若将以上几点策略与TCR-T共同使用或结合到TCR-T治疗的开发中,TCR-T治疗就有望成为对抗癌症的有力方法,尤其是针对实体瘤。通过结合免疫检查点抑制剂治疗[75]、新抗原疫苗、溶瘤病毒[69]、放疗等多种组合方法,可以显著改善癌症免疫治疗的现状,在临床前或早期临床试验中已经取得了一些令人振奋的结果,且此方法适用于多种类型的肿瘤,有望未来发展为一种广谱的抗癌治疗手段,同时,也为更多TCR-T产品的开发提供了新思路。随着全球机构逐年增长的研究投入,以及基因工程技术的不断进步与发展,相信TCR-T治疗在不久的将来可以带来更多癌症治疗领域的突破。
利益冲突声明:所有作者均声明不存在利益冲突。
[参考文献]
[1]MO Z M, DU P X, WANG G P, et al. The multi-purpose tool of tumor immunotherapy: gene-engineered T cells[J]. J Cancer, 2017, 8(9): 1690-1703.
[2]SCOTT L J. Osimertinib as first-line therapy in advanced NSCLC: a profile of its use[J]. Drugs Ther Perspect, 2018, 34(8): 351-357.
[3]WEBER E W, MAUS M V, MACKALL C L. The emerging landscape of immune cell therapies[J]. Cell, 2020, 181(1): 46-62.
[4]JIANG X T, XU J, LIU M F, et al. Adoptive CD8+ T cell therapy against cancer: challenges and opportunities[J]. Cancer Lett, 2019, 462: 23-32.
[5]SALTER A I, RAJAN A, KENNEDY J J, et al. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function[J]. Sci Signal, 2021, 14(697): eabe2606.
[6]XU Y Y, YANG Z Y, HORAN L H, et al. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release[J]. Cell Discov, 2018, 4: 62.
[7]GARBER K. Driving T-cell immunotherapy to solid tumors[J]. Nat Biotechnol, 2018, 36(3): 215-219.
[8]CHAPUIS A G, EGAN D N, BAR M, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant[J]. Nat Med, 2019, 25(7): 1064-1072.
[9]BLÜTHMANN H, KISIELOW P, UEMATSU Y, et al. T-cell-specific deletion of T-cell receptor transgenes allows functional rearrangement of endogenous alpha- and beta-genes[J]. Nature, 1988, 334(6178): 156-159.
[10]BUONAGURO L, TAGLIAMONTE M. Selecting target antigens for cancer vaccine development[J]. Vaccines (Basel), 2020, 8(4): 615.
[11]ZHANG J X, WANG L Y. The emerging world of TCR-T cell trials against cancer: a systematic review[J]. Technol Cancer Res Treat, 2019, 18: 1533033819831068.
[12]ZHAO Q J, JIANG Y, XIANG S X, et al. Engineered TCR-T cell immunotherapy in anticancer precision medicine: pros and cons[J]. Front Immunol, 2021, 12: 658753.
[13]HELLMAN L M, FOLEY K C, SINGH N K, et al. Improving T cell receptor on-target specificity via structure-guided design[J]. Mol Ther, 2019, 27(2): 300-313.
[14]TAWARA I, KAGEYAMA S, MIYAHARA Y, et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS[J]. Blood, 2017, 130(18): 1985-1994.
[15]ROBBINS P F, MORGAN R A, FELDMAN S A, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1[J]. J Clin Oncol, 2011, 29(7): 917-924.
[16]MORGAN R A, CHINNASAMY N, ABATE-DAGA D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy[J]. J Immunother, 2013, 36(2): 133-151.
[17]CAMERON B J, GERRY A B, DUKES J, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells[J]. Sci Transl Med, 2013, 5(197): 197ra103.
[18]YE B X, STARY C M, GAO Q P, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies[J]. J Immunol Res, 2017, 2017: 5210459.
[19] LIM W A, JUNE C H. The principles of engineering immune cells to treat cancer[J]. Cell, 2017, 168(4): 724-740.
[20] MOORE A R, ROSENBERG S C, MCCORMICK F, et al. RAStargeted therapies: is the undruggable drugged? [J]. Nat Rev Drug Discov, 2020, 19(8): 533-552.
[21] WANG Q J, YU Z Y, GRIFFITH K, et al. Identification of T-cell receptors targeting KRAS-mutated human tumors[J]. Cancer Immunol Res, 2016, 4(3): 204-214.
[22] MAOZ A, RENNERT G, GRUBER S B. T-cell transfer therapy targeting mutant KRAS[J]. N Engl J Med, 2017, 376(7): e11.
[23] DESAI J, GAN H, BARROW C, et al. Phase Ⅰ, open-label, dose-escalation/dose-expansion study of lifirafenib (BGB-283), an RAF family kinase inhibitor, in patients with solid tumors[J]. J Clin Oncol, 2020, 38(19): 2140-2150.
[24] HOOGEVEEN R C, ROBIDOUX M P, SCHWARZ T, et al. Phenotype and function of HBV-specific T cells is determined by the targeted epitope in addition to the stage of infection[J]. Gut, 2019, 68(5): 893-904.
[25] JIN B Y, CAMPBELL T E, DRAPER L M, et al. Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model[J]. JCI Insight, 2018, 3(8): e99488.
[26] NAGARSHETH N B, NORBERG S M, SINKOE A L, et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers[J]. Nat Med, 2021, 27(3): 419-425.
[27] DRAPER L M, KWONG M L M, GROS A, et al. Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6[J]. Clin Cancer Res, 2015, 21(19): 4431-4439.
[28] JOHNSON L A, MORGAN R A, DUDLEY M E, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen[J]. Blood, 2009, 114(3): 535-546.
[29] PARKHURST M R, YANG J C, LANGAN R C, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis[J]. Mol Ther, 2011, 19(3): 620-626.
[30] LINETTE G P, STADTMAUER E A, MAUS M V, et al. Cardiovascular toxicity and titin cross-reactivity of affinityenhanced T cells in myeloma and melanoma[J]. Blood, 2013, 122(6): 863-871.
[31] HONG D S, VAN TINE B A, OLSZANSKI A J, et al. Phase Ⅰ dose escalation and expansion trial to assess the safety and efficacy of ADP-A2M4 SPEAR T cells in advanced solid tumors[J]. J Clin Oncol, 2020, 38(15_suppl): 102.
[32] LAM V K, HONG D S, HEYMACH J, et al. Initial safety assessment of MAGE-A10c796 TCR T-cells in two clinical trials[J]. J Clin Oncol, 2018, 36(15_suppl): 3056.
[33] DORAN S L, STEVANOVIĆ S, ADHIKARY S, et al. T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: a first-in-human, phase Ⅰ/Ⅱ study[J]. J Clin Oncol, 2019, 37(30): 2759-2768.
[34] DAVIS J L, THEORET M R, ZHENG Z L, et al. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials[J]. Clin Cancer Res, 2010, 16(23): 5852-5861.
[35] KERNEL N N I. TCR-redirected T cells therapy in patient with HBV related HCC[J]. Case Med Res, 2019.[Epub ahead of print].
[36] SPEAR T T, EVAVOLD B D, BAKER B M, et al. Understanding TCR affinity, antigen specificity, and cross-reactivity to improve TCR gene-modified T cells for cancer immunotherapy[J]. Cancer Immunol Immunother, 2019, 68(11): 1881-1889.
[37] SANDERSON J P, CROWLEY D J, WIEDERMANN G E, et al. Preclinical evaluation of an affinity-enhanced MAGEA4-specific T-cell receptor for adoptive T-cell therapy[J]. Oncoimmunology, 2020, 9(1): 1682381.
[38] KHONG H T, WANG Q J, ROSENBERG S A. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression[J]. J Immunother, 2004, 27(3): 184-190.
[39] NATHAN P, HASSEL J C, RUTKOWSKI P, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma[J]. N Engl J Med, 2021, 385(13): 1196-1206.
[40] WALKER A J, MAJZNER R G, ZHANG L, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase[J]. Mol Ther, 2017, 25(9): 2189-2201.
[41] ROBINSON J, HALLIWELL J A, HAYHURST J D, et al. The IPD and IMGT/HLA database: allele variant databases[J]. Nucleic Acids Res, 2015, 43(Database issue): D423-D431.
[42] GONZALEZ-GALARZA F F, MCCABE A, SANTOS E J M D, et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools[J]. Nucleic Acids Res, 2020, 48(D1): D783-D788.
[43] GARETTO S, SARDI C, MARTINI E, et al. Tailored chemokine receptor modification improves homing of adoptive therapy T cells in a spontaneous tumor model[J]. Oncotarget, 2016, 7(28): 43010-43026.
[44] IDORN M, SKADBORG S K, KELLERMANN L, et al. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model[J]. Oncoimmunology, 2018, 7(8): e1450715.
[45] HU J M, SUN C, BERNATCHEZ C, et al. T-cell homing therapy for reducing regulatory T cells and preserving effector T-cell function in large solid tumors[J]. Clin Cancer Res, 2018, 24(12): 2920-2934.
[46] ADACHI K, KANO Y, NAGAI T, et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor[J]. Nat Biotechnol, 2018, 36(4): 346-351.
[47] FRAIETTA J A, LACEY S F, ORLANDO E J, et al. Author correction: determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia[J]. Nat Med, 2021, 27(3): 561.
[48]BECHMAN N, MAHER J. Lymphodepletion strategies to potentiate adoptive T-cell immunotherapy-what are we doing; where are we going?[J]. Expert Opin Biol Ther, 2021, 21(5): 627-637.
[49]CHEN J, SUN H W, YANG Y Y, et al. Reprogramming immunosuppressive myeloid cells by activated T cells promotes the response to anti-PD-1 therapy in colorectal cancer[J]. Signal Transduct Target Ther, 2021, 6(1): 4.
[50]KIM J, KANG S, KIM K W, et al. Nanoparticle delivery of recombinant IL-2 (BALLkine-2) achieves durable tumor control with less systemic adverse effects in cancer immunotherapy[J]. Biomaterials, 2022, 280: 121257.
[51]SAKAI T, TERAKURA S, MIYAO K, et al. Artificial T cell adaptor molecule-transduced TCR-T cells demonstrated improved proliferation only when transduced in a higher intensity[J]. Mol Ther Oncolytics, 2020, 18: 613-622.
[52]ROTH T L, LI P J, BLAESCHKE F, et al. Pooled knockin targeting for genome engineering of cellular immunotherapies[J]. Cell, 2020, 181(3): 728-744.e21.
[53]MARTINEZ M, MOON E K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment[J]. Front Immunol, 2019, 10: 128.
[54]NALAWADE S A, SHAFER P, BAJGAIN P, et al. Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer[J]. J Immunother Cancer, 2021, 9(11): e003237.
[55]LIN S H, CHENG L, YE W, et al. Chimeric CTLA4-CD28-CD3z T cells potentiate antitumor activity against CD80/CD86-positive B cell malignancies[J]. Front Immunol, 2021, 12: 642528.
[56]NEWICK K, O'BRIEN S, MOON E, et al. CAR T cell therapy for solid tumors[J]. Annu Rev Med, 2017, 68: 139-152.
[57]D’ALOIA M M, ZIZZARI I G, SACCHETTI B, et al. CAR-T cells: the long and winding road to solid tumors[J]. Cell Death Dis, 2018, 9(3): 282.
[58]ZHANG B L, QIN D Y, MO Z M, et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors[J]. Sci China Life Sci, 2016, 59(4): 340-348.
[59]YE B X, SMERIN D, GAO Q P, et al. High-throughput sequencing of the immune repertoire in oncology: applications for clinical diagnosis, monitoring, and immunotherapies[J]. Cancer Lett, 2018, 416: 42-56.
[60]CHEN F J, ZOU Z Y, DU J, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors[J]. J Clin Invest, 2019, 129(5): 2056-2070.
[61]JOGLEKAR A V, LEONARD M T, JEPPSON J D, et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors[J]. Nat Methods, 2019, 16(2): 191-198.
[62]CHEN X J, PONCETTE L, BLANKENSTEIN T. Human TCR-MHC coevolution after divergence from mice includes increased nontemplate-encoded CDR3 diversity[J]. J Exp Med, 2017, 214(11): 3417-3433.
[63]KRSHNAN L, PARK S, IM W, et al. A conserved αβ transmembrane interface forms the core of a compact T-cell receptor-CD3 structure within the membrane[J]. Proc Natl Acad Sci U S A, 2016, 113(43): E6649-E6658.
[64]KNIES D, KLOBUCH S, XUE S A, et al. An optimized single chain TCR scaffold relying on the assembly with the native CD3-complex prevents residual mispairing with endogenous TCRs in human T-cells[J]. Oncotarget, 2016, 7(16): 21199-21221.
[65]ARMISTEAD P M. Cellular therapy against public neoantigens[J]. J Clin Invest, 2019, 129(2): 506-508.
[66]LU Y C, ZHENG Z L, LOWERY F J, et al. Direct identification of neoantigen-specific TCRs from tumor specimens by high-throughput single-cell sequencing[J]. J Immunother Cancer, 2021, 9(7): e002595.
[67]LI S R, HUO F Y, WANG H Q, et al. Recent advances in porous nanomaterials-based drug delivery systems for cancer immunotherapy[J]. J Nanobiotechnology, 2022, 20(1): 277.
[68]CADILHA B L, BENMEBAREK M R, DORMAN K, et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors[J]. Sci Adv, 2021, 7(24): eabi5781.
[69]EVGIN L, KOTTKE T, TONNE J, et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice[J]. Sci Transl Med, 2022, 14(640): eabn2231.
[70]PARKHURST M, GROS A, PASETTO A, et al. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression[J]. Clin Cancer Res, 2017, 23(10): 2491-2505.
[71]MARCU A, BICHMANN L, KUCHENBECKER L, et al. HLA Ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy[J]. J Immunother Cancer, 2021, 9(4): e002071.
[72]HU Z D, ZHU L Y, WANG J, et al. Immune signature of enhanced functional avidity CD8+ T cells in vivo induced by vaccinia vectored vaccine[J]. Sci Rep, 2017, 7: 41558.
[73]ALBA J, D‘ABRAMO M. The full model of the pMHC-TCR-CD3 complex: a structural and dynamical characterization of bound and unbound states[J]. Cells, 2022, 11(4): 668.
[74]HE W H, CAO Z L, MAO F F, et al. Modification of three amino acids in sodium taurocholate cotransporting polypeptide renders mice susceptible to infection with hepatitis D virus in vivo[J]. J Virol, 2016, 90(19): 8866-8874.
[75]CAO Y Q, LU W Y, SUN R, et al. Anti-CD19 chimeric antigen receptor T cells in combination with nivolumab are safe and effective against relapsed/refractory B-cell non-Hodgkin lymphoma[J]. Front Oncol, 2019, 9: 767.

