基底神经节和丘脑(中央灰质)的双侧病变图解总结 | 一文掌握
时间:2022-12-28 18:00:08 热度:37.1℃ 作者:网络
huihui的学习笔记
基底神经节和丘脑是成对的灰质结构,在大脑半球深部,通常被称为 "中央灰质"。它们参与调节自主神经、运动、感觉、边缘和内分泌功能,因此,它们的代谢需求超过了静止状态下的大脑皮层。基底神经节病变通常会导致运动障碍,但其作用超出了锥体外系,延伸到记忆、情感和其他认知功能。丘脑是复杂的中枢,主要接受皮质下的感觉和运动输入,投射到大脑皮层和纹状体。丘脑病变可引起慢性疼痛、感觉丧失、失忆、肌张力障碍和其他疾病。
由于名称和定义的多样性、复杂的相互联系和相互作用,术语可能令人困惑。基底神经节的组成部分和命名方法取决于考虑这些复杂结构的哪个方面(生物化学、胚胎学或功能)。在本综述中,"基底神经节 "一词是在最严格的意义上使用的(端脑基底神经节),指的是纹状体(包括尾状核、壳核和苍白球)。黑质和丘脑下核分别起源于间脑和中脑,在本评论中不被视为基底神经节的一部分。从胚胎学和功能上看,尾状核和壳核形成一个单元,即纹状体。豆状核是壳核和苍白球的一个描述性统称,壳核和苍白球被一薄层白质,即外侧髓质层所分隔。
丘脑位于第三脑室的边缘,起源于胚胎期的脑膜,由灰质核团和白质束组成。胚胎起源的差异也反映在动脉供血上:基底神经节由大脑前动脉的穿孔分支供应,包括Heubner's动脉、大脑中动脉和前脉络膜动脉,它们都源于颈内动脉;大脑后动脉的穿孔分支和后交通动脉供应丘脑。丘脑静脉收集基底神经节和丘脑的静脉回流,然后血液流经大脑内静脉和Rosenthal基底静脉,形成Galen静脉,并进入直窦。
磁共振成像(MRI)是评估涉及深层灰质核的病变的首选方式,这是因为与计算机断层扫描(CT)相比,其对比度更强。尽管如此,CT作为检测钙化的参考标准具有明显的作用,而敏感性加权磁共振成像(SW-MRI)的准确性略低。然而,当没有CT或只能进行一次检查时,SW-MRI,尤其是该序列的相位信息,是CT检测钙化的有效替代方法。
在CT上,基底神经节和丘脑通常与大脑皮层呈等密度。在T2加权和FLAIR图像上,丘脑、壳核和尾状核的信号强度与大脑皮层相似,而苍白球则由于铁的沉积而出现轻微低密度。苍白球T1信号强度较高主要是由于髓磷脂含量较高,但也与钙沉积有关。T1信号强度随着钙浓度达到30%而增加,但随着钙浓度的增加而降低。在目前使用的临床MRI扫描仪上(场强为1.5–3 T),将苍白球分为内部和外部的内部髓质层没有很好地显示出来。基底神经节和丘脑在注射造影剂后没有增强,在表观弥散系数(ADC)图上也没有显示弥散性的改变。在SW-MRI上,由于钙和铁的沉积,苍白球显示出更明显的低密度。所有深层灰质结构都会出现随年龄增长而增加的矿化现象和体积损失。
疾病
根据影像学检查中病变的主要位置(基底节区、丘脑或两者都有),这些疾病被分为三部分,并根据病理类型进一步细分(主要是遗传代谢/遗传、获得性代谢/毒性、炎症和感染、血管和缺血、肿瘤)。本文最后的概要图由多个裁剪的轴向图像组成,显示了涉及基底节区和丘脑的最典型的疾病模式。
影响基底神经节的主要疾病
一、遗传性代谢/基因
亚急性坏死性脑病(Leigh综合征)
亚急性坏死性脑病(leigh综合征)是一种进行性线粒体神经退行性疾病,预后严重,常出现在婴儿期或儿童早期。这种最常见的线粒体疾病是基因异质性的,在超过75个基因中发现了突变。受影响的婴儿表现为进食困难,精神运动障碍和共济失调,最常见于代谢应激期间或之后。动脉血液和/或脑脊液中乳酸水平升高(也可在脑磁共振波谱中发现)是诊断的线索。
MRI显示,在壳核(特征性但不总是存在)和/或脑干(中脑和髓质,包括导水管周围灰质)双侧对称的高T2信号区(图1、2和59),由于细胞毒性水肿,在急性期会出现弥散性受限。丘脑下核、黑质、尾状核、苍白球、丘脑背内侧核和小脑齿状核也可能受累。这些MRI信号改变反映了受影响大脑结构的海绵状改变和空泡化。
在其他线粒体疾病、威尔逊病、缺氧缺血性脑病和中毒原因中也可看到类似的结果。然而,双侧对称T2高信号累及多个脑干结构和基底神经节的神经问题的儿童应提示考虑Leigh综合征
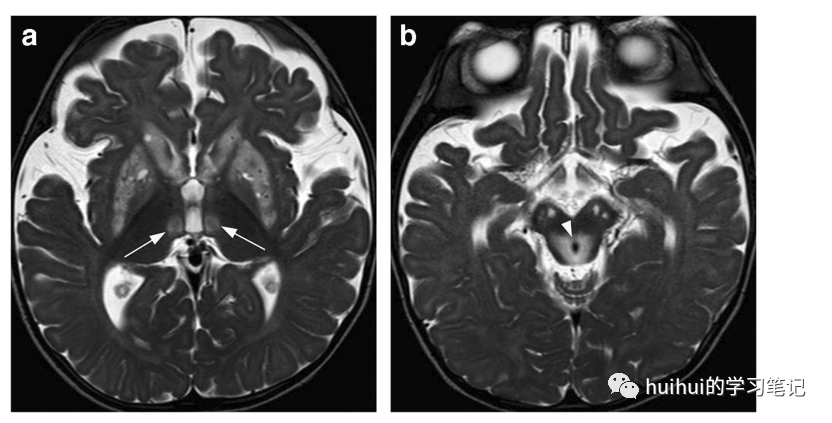
图1:Leigh综合症。6岁女童Leigh综合征急性期。基底神经节水平轴向t2加权图像显示双侧对称纹状体高信号和局灶性丘脑内侧病变(箭头)。苍白球信号强度稍增高。b:中脑轴向t2加权图像显示顶盖、导水管周围灰质(箭头)和双侧黑质高信号。在回声时间为144 ms的MR光谱中,检测到乳酸在1.33 ppm处呈倒双峰(未显示).
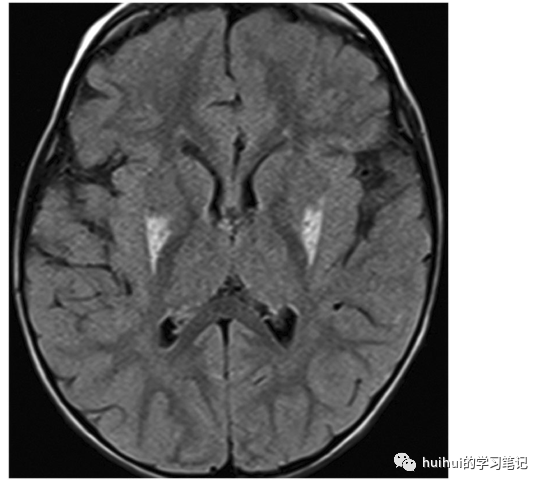
图2:Leigh综合症。一名患有Leigh综合征的4岁男孩。轴位FLAIR像显示双侧硬膜背侧对称高信号。有其他对称脑干病变(未显示)
戊二酸尿症Ⅰ型
戊二酸尿1型是一种常染色体隐性代谢疾病,常见于婴儿。该病经常在感染或免疫接种后突然发病,出现局灶性或全身性发作。随后症状演变为精神运动倒退和肌张力障碍或舞蹈性手足徐动。大头畸形症通常在出生时出现。
MRI显示特征性额颞萎缩伴Sylvian裂扩大(“蝙蝠翼”扩张),硬膜下水肿和髓鞘形成迟缓(图3,4和59)。尾状核和壳核T2信号增强,急性期肿胀,弥漫性降低,随时间推移演变为萎缩。在某些病例中可见沿侧脑室的融合性白质信号改变和室管膜下结节(图3b)。由于硬膜下水肿的存在,这种情况可能是一种非意外创伤的模拟。然而,特征性相关异常应提醒放射科医生代谢疾病的可能性。

图3:戊二酸尿症1型。14岁男性戊二酸尿症1型。a:轴位t2加权像上,双侧对称后壳核后部高信号(箭头)。b:在冠状t2加权图像上可以更好地看到额颞顶叶汇合的高信号区(白色箭头),并显示沿额角上方的室管膜下结节(黑色箭头)
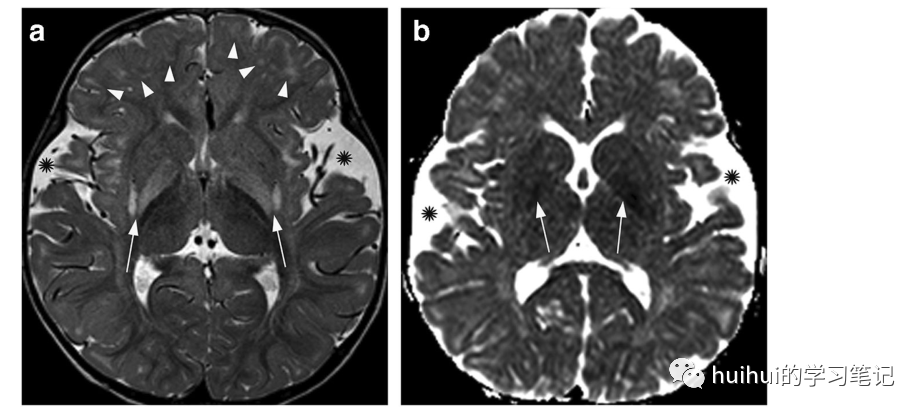
图4:戊二酸尿症1型。一个20个月大的男孩戊二酸尿症1型。轴位t2加权图像显示皮层下迟发髓鞘形成区,主要是额叶(箭头),以及更明显的双侧对称苍白球和壳核后部高信号区(箭头)。b ADC图显示豆状核(箭头)信号强度较低,与弥散率降低一致。双侧扩大的外侧裂(*),图a更明显。
Wilson病
肝豆状核变性(WD)是一种常染色体隐性遗传病,由ATP7B基因突变引起,导致铜在肝脏中的转运和积累错误,并沉积在包括大脑在内的其他部位。诊断通常是由低铜蓝蛋白水平和虹膜周围Kayser-Fleischer环的存在,由高尿铜支持,并由基因检测确认。锥体外系和行为症状通常开始由20-30岁开始出现;但是,它可能在老年人和幼儿中表现出来,在t2加权和FLAIR图像上,有双侧对称的深灰质信号改变,通常为同心层流高信号。壳核(特别是沿外缘)的高T2信号最常被观察到(图5a和图59),其次是尾状核、苍白球、丘脑(保留内侧部分)和脑干的变化。弥漫率降低有时可能出现在非常早期的阶段;然而,扩散增加更为典型。对称T1高强度苍白球是常见的(图5b和59),可能是由肝衰竭引起的。中脑高强度,保留红核、部分黑质和上丘,产生“大熊猫脸”征象,这实际上是WD的典型症状,但仅在少数患者中存在。
异常信号改变随着铜螯合治疗的反应而消退。类似的神经影像学表现可见于Leigh病、甲醇中毒和代谢性酸中毒。然而,双侧基底节区在MRI上伴或不伴丘脑或脑干病变在适当的临床环境下高度提示WD。
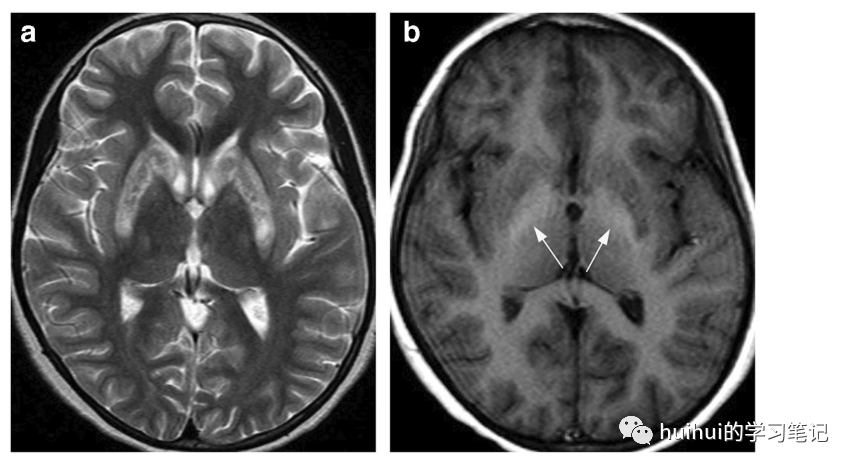
图5:Wilson病。一名六岁女童,头痛,震颤,肝功能衰竭。轴位t2加权像显示双侧对称的壳核和尾状核高信号,周围更突出。双侧苍白球和外侧丘脑的信号强度有细微增加。b:稍低水平轴位t1加权图像显示双侧壳核信号强度轻度降低,苍白球信号强度高(箭头).
亨廷顿病
亨廷顿病(HD)是一种常染色体显性神经退行性疾病,由编码亨廷顿蛋白的基因突变(CAG重复序列的延长,编码谷氨酰胺)引起。突变的亨廷顿蛋白聚集在轴突末梢,并导致神经元细胞死亡,这在纹状体的棘状氨基丁酸能神经元中最为明显。该疾病的特征是运动、认知和精神症状的三联征,在15-20年的时间里表现出不可逆的进展病程。发病的平均年龄为40岁,尽管HD病例已在2至87岁的患者中确诊。HD在成人中的典型发病形式表现为多动性,而青少年表现为低动性运动障碍。
HD的特征性影像学表现为双侧纹状体(尾状核和壳核)萎缩,尤其累及尾状核的头部。这导致相邻的前角对称增大,其横向轮廓变平,形成“盒子”外观(图6和图59)。尾状叶之间的距离超过20毫米,而正常值为10到14毫米。由于铁沉积导致的轻度相关T2低强度可在成人疾病中注意到。另一方面,在幼年期起病的HD中,尾状核和壳核中经常可见高T2信号(图7)。随着疾病的进展还可出现弥漫性脑萎缩伴白质体积减少,额叶最明显。体积MRI提示基因突变的携带者在症状出现的数年前就开始出现微小但进行性的纹状体和脑白质萎缩。 图6:亨廷顿疾病。36岁男性患者,有亨廷顿病和舞蹈病家族史。冠状位造影后t1加权像显示尾状核头严重萎缩,侧脑室额角呈“盒子状”外观(箭头)。额叶也可见萎缩。
图6:亨廷顿疾病。36岁男性患者,有亨廷顿病和舞蹈病家族史。冠状位造影后t1加权像显示尾状核头严重萎缩,侧脑室额角呈“盒子状”外观(箭头)。额叶也可见萎缩。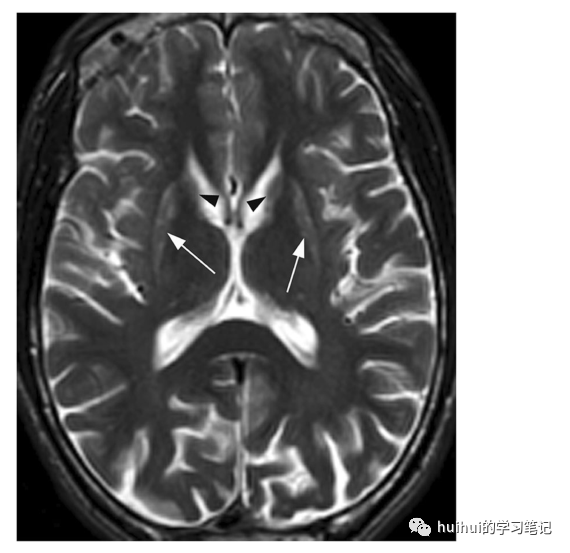
图7:亨廷顿疾病。一名17岁的青少年患者。轴向t2加权图像显示双侧尾状核(箭头)和壳核(箭头)变小。相关的高信号在壳核中更为明显
伴有脑铁积累的神经退行性变
脑铁积累性神经退行性变(NBIA)是一组以基底神经节铁异常积累为特征的遗传性神经系统疾病。疾病发病时间从婴儿期到成年期不等,进展可快可慢,长期稳定。这些患者表现为肌张力障碍、痉挛、帕金森症和神经精神障碍,MRI显示铁沉积。全身性脑和小脑萎缩也经常出现。
在这些疾病中,铁的分布模式各不相同,有额外的特征有助于鉴别诊断。“虎眼”征指的是t2加权图像上苍白球的低强度和中央高强度(图8和图59),几乎是泛酸激酶相关神经退行性变(PKAN,以前称为Hallervorden-Spatz病)的典型症状,这是最常见的疾病(高达50%的NBIA病例)。此外,在线粒体膜蛋白相关神经退行性变(MPAN)中,部分患者内、外苍白球之间的内侧髓片出现T2高强度条纹。最后,尾状核和壳核的囊性改变和空泡化是神经铁蛋白病所特有的。
请注意,在威尔逊病、非典型帕金森症或有机磷中毒中也可观察到“虎眼”征,甚至在3T核磁中,正常人也可出现。另一方面,核磁共振成像特征可能先于NBIA障碍患者的症状。
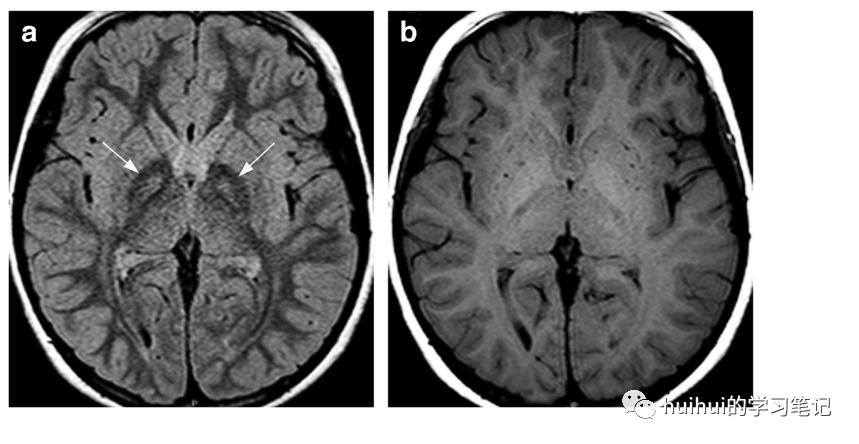 图8:PKAN。一PKAN。一名患有泛酸激酶相关神经变性的8岁男孩。a:FLAIR图像上双侧苍白球前内侧对称高信号灶被低信号包围(箭头),表现为典型的“老虎眼”征。b:t1加权图像上有相应的轻度高信号
图8:PKAN。一PKAN。一名患有泛酸激酶相关神经变性的8岁男孩。a:FLAIR图像上双侧苍白球前内侧对称高信号灶被低信号包围(箭头),表现为典型的“老虎眼”征。b:t1加权图像上有相应的轻度高信号
二、获得代谢/毒物
一氧化碳中毒
在世界各地,由于碳基燃料的不完全燃烧,暴露于一氧化碳(CO)是导致人类中毒的最常见原因。吸入一氧化碳后,与血红蛋白结合的亲和力比氧强250倍,从而降低携氧能力,导致随后的组织缺氧。此外,CO阻止细胞对氧气的摄入,从而损害线粒体呼吸功能,促进氧自由基的形成。一氧化碳中毒的神经症状不仅可立即出现,而且可在初步复苏成功后2至6周作为迟发性神经后遗症出现。
CO中毒的影像学主要为双侧局灶性苍白球病变,在CT上呈低密度,在T2、FLAIR和DWI上呈高信号(图9、图10和图59)是非常典型的影像学表现,在急性期可见相应的弥散降低。部分患者在梯度回声(GRE) T2*加权图像/SW-MRI上出现中心或外周低信号区,偶有相应的T1高信号,提示出血。在急性期,也可看到斑片状和/或周围造影剂增强。早期常见的影像学还有双侧脑白质T2和DWI高信号。偶尔可累及壳核、尾状核、丘脑、海马、小脑和黑质,通常是一些为较细微的病变。另一方面,甲醇中毒和缺氧缺血性脑病(HIE)患者可出现双侧苍白球病变的孤立异常。有趣的是,大脑白质信号改变可能实际上代表了CO中毒最常见的MRI发现,而不是苍白球的特征性病变。白质病变也被认为是慢性症状的主要原因。
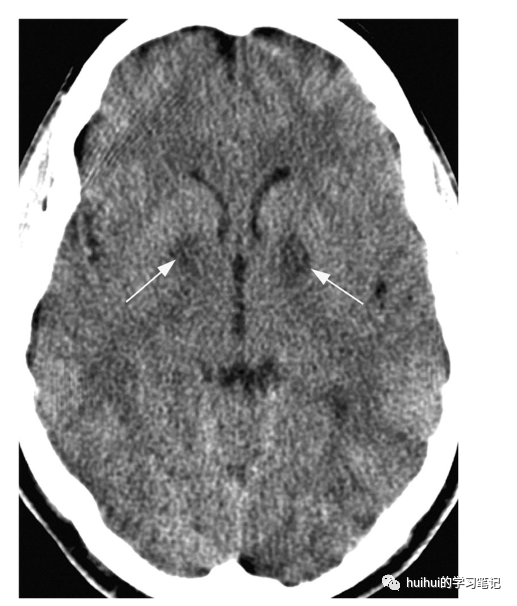
图9:一氧化碳中毒。37岁火灾受害者头痛,恶心,呕吐。轴位CT显示双侧苍白球内局灶性低密度病变(箭头)
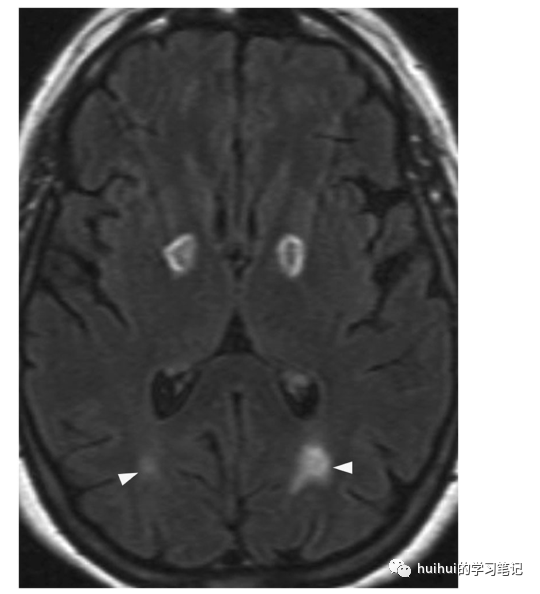
图10:CO中毒。加热器故障导致急性一氧化碳中毒。轴位FLAIR像显示双侧苍白球椭圆形高信号病变,中心低信号(提示小出血)。也有白质高强度(箭头),在半卵圆体更广泛(未显示)
胆红素脑病(核状脑病)
核黄疸是一种获得性胆红素脑病的慢性形式,在新生儿期胆红素脑病急性期存活的婴儿中发病,发病时间长达数月至数年。慢性期的神经影像学特征是苍白球(图11和图59)和丘脑下核中存在双侧对称高T2信号。
黑质和小脑齿状核内也可见T2信号增加,T1信号变化不定。另一方面,急性至亚急性期MRI特征性表现为苍白球和丘脑下核T1信号增加。与缺氧缺血性脑病相比,无T2高强度或弥散异常,丘脑和壳核外观正常。在足月新生儿中,一个关键的区别是正常的髓鞘形成,因为苍白球和丘脑下核在t1加权序列上自发高强度。值得注意的是,3D GRE t1加权图像在3t时显示在这些正常髓鞘形成的区域有特殊的高信号倾向。
图11:核黄疸。一名4岁男孩患有慢性胆红素脑病。t2加权图像显示苍白球对称高信号。对称的增加信号也出现在齿状核和丘脑下核(未显示)。
慢性肝性脑病/锰毒性
慢性肝性脑病(CHE)最常发生在肝硬化、门静脉高压或门静脉系统分流的情况下。它包括一系列由肝功能障碍引起的神经精神异常。
双侧对称苍白球t1加权图像高信号是CHE患者的特征性表现(图12和图59),在CT或t2加权MRI上无相应异常。双侧小脑齿状核、黑质、丘脑下核和顶盖也可见高T1信号。锰(Mn)的积累被认为是T1信号改变的原因。肝硬化患者血清中锰的浓度是健康人的三倍,这是由于肝脏排除不充分。弥漫性白质T2高信号优先累及皮质脊髓束和皮层下局灶性高T2病变也可出现。
肝功能的恢复,如肝移植,导致MRI异常的改善,包括T1高强度,白质T2信号增加,以及光谱结果(肌肌醇水平降低,谷氨酰胺/谷氨酸复合物升高)和恢复正常外观。值得注意的是,在长期全肠外营养、克罗恩病和长期使用质子泵抑制剂治疗胃溃疡的患者中可以看到锰沉积。

图12:慢性肝衰竭/锰毒性。49岁女性,已知肝硬化,新发震颤。轴位t1加权像上可见苍白球对称高信号。增加的信号延伸到中脑被盖和齿状核(未显示)。内囊内T2信号强度也增加(未显示)
钆沉积
在多次增强MRI扫描后,主要使用线性钆基造影剂(GBCA),苍白球和小脑齿状核呈对称T1高强度,常伴有钆沉积。这种现象在2014年首次被报道,目前还不知道Gd沉积导致的神经、神经心理学或其他临床后果。MRI表现可能无法与慢性肝性脑病或其他锰积累原因的患者区分开来。
甲醇中毒
甲醇中毒通常发生在口服工业液体或饮用受污染的酒精饮料后,通常发生在集体中毒中。摄入后最明显的急性症状是视觉障碍。其他症状延迟至24小时,因为甲醇在肝脏中被乙醇脱氢酶代谢为甲酸,导致严重的代谢性酸中毒。治疗的主要方法是静脉注射乙醇,因为乙醇对酒精脱氢酶有较高的亲和力。甲醇中毒有很高的死亡率,并导致幸存者长期的视觉后遗症和严重的脑损伤。
在摄入甲醇24 - 36小时后可见典型的出血性或非出血性双侧对称壳核病变。壳膜在CT上呈低密度,而在t2加权MR图像上呈高强度(图13和图59),通常T1高强度反映出血。在急性期发现弥散率降低,可能有(通常是周围)造影增强。尾状核和苍白球以及皮层也可能受累。双侧壳核出血,特别是伴随相关的额叶和岛叶白质病变(图13和59),几乎是急性甲醇中毒的典型症状,而在尿毒症脑病和其他代谢性酸中毒患者中可以看到孤立的壳核(和尾状核)对称受累
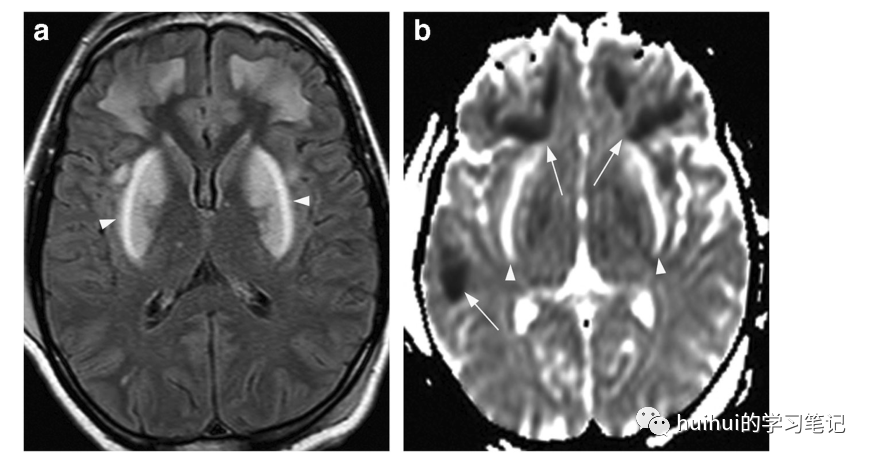
图13:甲醇中毒。一名31岁的男子在饮用自家酿造的烈酒几天后。轴位FLAIR像显示双侧对称高信号,硬膜和额叶白质轻度肿胀。沿硬膜外侧缘(箭头)信号改变明显,并有少量额外小的高信号区。b稍低水平ADC图显示硬膜弥散率高(箭头),受影响的白质弥散减低(箭头),主要位于额叶。
尿毒症脑病和代谢性酸中毒
尿毒症脑病是一种罕见的终末期肾功能衰竭合并颅内尿毒症毒素积聚的并发症。
在这些患者中,MRI经常在t2加权和FLAIR图像上显示双侧壳核和苍白球对称肿胀。此外,有一个更亮的信号强度的边缘,从壳核后部沿外膜向外延伸,而其内侧臂沿内膜延伸,然后在前方分叉成两个强度稍低的分支,分别对应于外髓层和内髓层(图14a和59)。由此可见,豆状核被划分为壳核、内苍白球和外苍白球三部分,形似叉子。因此,这种影像学特征被命名为“豆状核叉子征”。ADC图显示外围明亮信号沿“分叉”扩散率增加,中间出现不同的外观,经常包含扩散率降低的暗区(图14b)。没有相关的造影增强,尾状核也可能受累。肾移植或肾透析可改善症状和影像学表现。慢形叉征可在代谢性酸中毒和糖尿病无酸中毒患者中发现,如二甲双胍相关脑病。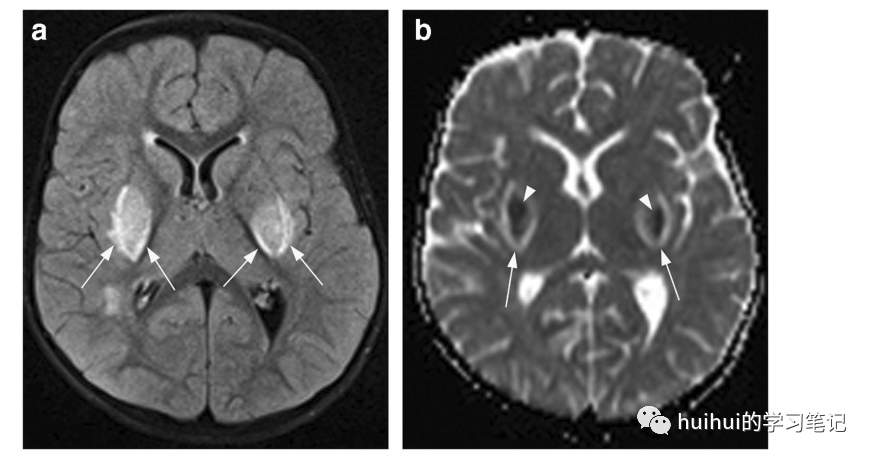
图14:尿毒症脑病/代谢性酸中毒。1例2岁女童因呕吐和腹泻入院1周。实验室结果证实溶血性尿毒症综合征。轴位FLAIR像显示双侧中央和后壳膜对称肿胀和信号增加,沿内侧边缘(箭头)有更明显的高强度边缘,这是尿毒症脑病和代谢性酸中毒的特征性“慢形叉”征象。还有一个小的白质信号改变。b病变在相应的ADC图上显示中心减少(箭头)和外围(沿壳膜内侧和外侧臂)扩散性增加(箭头)
低血糖
血糖水平突然下降(如糖尿病患者或外源性给胰岛素)可引起昏迷,但大多数患者在输注葡萄糖后恢复意识。低血糖脑病可在重度低血糖时发生,并可造成不可逆的脑损伤,导致持续的植物人状态或死亡。
MRI上可能出现三种类型的异常:选择性白质受累、选择性灰质受累或两者兼有。此外,病变可以是弥漫性的,也可以是局灶性的,可以是双侧的,不对称的,甚至是单侧的。据报道,大脑皮层、基底神经节、海马、皮质下白质、内囊后肢和胼胝体压部均有异常,而丘脑、小脑和脑干除外。严重低血糖的早期变化是由于细胞肿胀和细胞毒性水肿造成的弥散降低。异常的程度取决于低血糖的严重程度和持续时间,弥漫性和广泛性DWI病变预示着较差的神经预后。丘脑不受累是其与缺氧相关脑病的鉴别,在缺氧中,丘脑最常受影响(图15和59)。
短暂性低血糖在围生期新生儿中较为常见。异常影像通常是双侧顶叶和枕叶白质和灰质的受累。
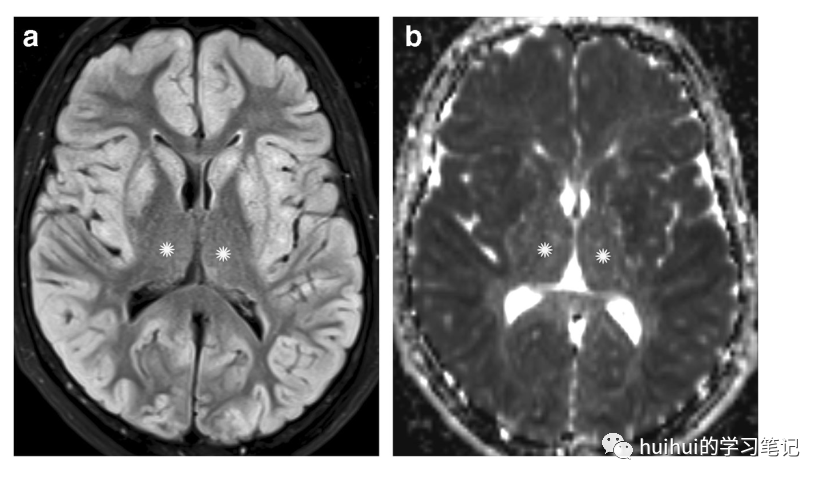
图15:低血糖。一名77岁的糖尿病女性,因胰岛素过量自杀未遂,导致低血糖昏迷。a:在轴向FLAIR图像上,全部大脑皮层和纹状体显示双侧高信号。b对应的ADC图显示受累灰质扩散率降低。苍白球和丘脑(*)被保留
高血糖
描述临床表现的非酮症性高血糖症伴半舞蹈症的病理过程在(控制不良)糖尿病患者中常见,更常见于亚裔老年女性。然而,在包括青少年在内的年轻人中,单侧肢体舞蹈和弹道运动(突然的抖动?)的突然发作可能是1型糖尿病的最初和唯一表现。
CT显示壳核高密度,一般位于症状的对侧。与所有其他代谢或毒性疾病相比,高血糖的病灶在绝大多数病例中具有特征性和单侧性(在罕见的双侧病例中通常显著不对称)。MRI显示相应的核壳核T1异常高信号,伴或不伴尾状核和苍白球受累(图16和图59)。病变部位在DWI、ADC、gret2 *加权(或SWMRI)和T2加权图像上表现不一。影像学特征通常是可逆的,尽管它们滞后于临床改善。
(译者:临床上遇见很多老年病人,怀疑脑梗死,一查血糖20mmol以上,这种需要警惕高血糖相关脑病,T1出现高信号不单单要怀疑梗死后渗血,还应怀疑高血糖相关病变)
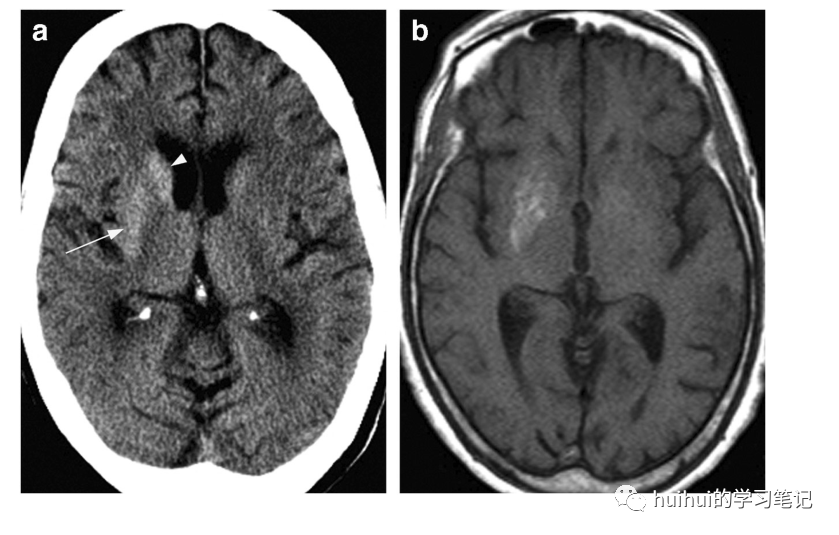
图16:高血糖症。一名79岁的女性,表现为左侧偏侧舞蹈病。实验室发现证实了这名已知糖尿病患者的非酮症高血糖症。轴向CT图像显示整个右壳核(箭头)和尾状头部(箭头)高度衰减,无扩张。b MRI显示T1加权成像显示右壳核高信号。
三、炎症/感染
自身免疫性脑炎
自身免疫性脑炎是指一组针对几种神经元抗原的免疫性病变,有或没有潜在的恶性肿瘤。在第一种情况下,抗体通常是针对细胞内抗原的,而在第二种情况下,抗体是针对表面受体的。无论病因或抗体情况如何,对边缘系统的抗原有明显的偏向性。然而,针对基底神经节的抗原的实体也被描述过。临床表现通常是渐进性,在几周内不断发展,精神症状很常见,同时还有运动障碍和癫痫发作。
抗CV2/CRMP5(塌陷素反应介导蛋白5 Collapsin response mediator protein 5 )脑炎,经常与肺癌和胸腺瘤有关,典型的表现为舞蹈症和行为障碍。在一些病人中,MRI显示T2加权和FLAIR图像上纹状体(壳核和尾状核)和屏状核有显著的高信号(图17和59),没有弥散异常,这有助于与克雅氏病相鉴别。抗CV2脑炎是一种排除性诊断,鉴别诊断应包括各种中毒和代谢性酸中毒。
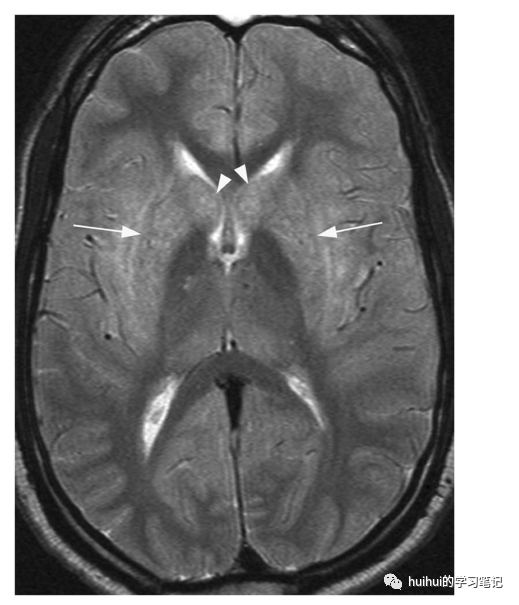
图17 自身免疫性脑炎。一名46岁的男性,有步态不稳、行为紊乱和舞蹈症。轴向T2加权图像显示双侧对称性高信号和轻度肿胀,中心位于壳核(箭头)和尾状核(箭头)。在扣带回和丘脑后部有轻微的对称性信号强度增加。抗CV2/CRMP5抗体检测为阳性,最终诊断为转移性肺癌
隐球菌病
隐球菌中枢神经系统感染的表现是可变的,包括胶状假囊、肉芽肿(隐球菌)和脑膜脑炎。影像学检查,特别是CT,可能是正常的或仅显示脑室肿大。病原体通过血管周围的空间从基底贮水池延伸到大脑,形成充满真菌和其胶状囊状物质的囊状结构。这些特征性的胶状假囊肿是分界清楚的病变,主要位于基底节,大小从几毫米到几厘米不等。
假性囊肿在CT以及T2和T1加权MRI上无强化,呈CSF样,但在FLAIR图像上呈高信号(图18a和59)。在免疫抑制的病人中发现多个囊肿/扩张的血管周围间隙,高度怀疑是隐球菌感染。另一方面,隐球菌是T2高信号的结节,通常位于深部灰质,显示轻度对比剂增强(图18b),可能有周围水肿。虽然胶质假囊和隐球菌是HIV感染者最常见的发现,但放射学上的脑膜炎是HIV阴性者隐球菌脑膜脑炎最主要的MRI异常现象。
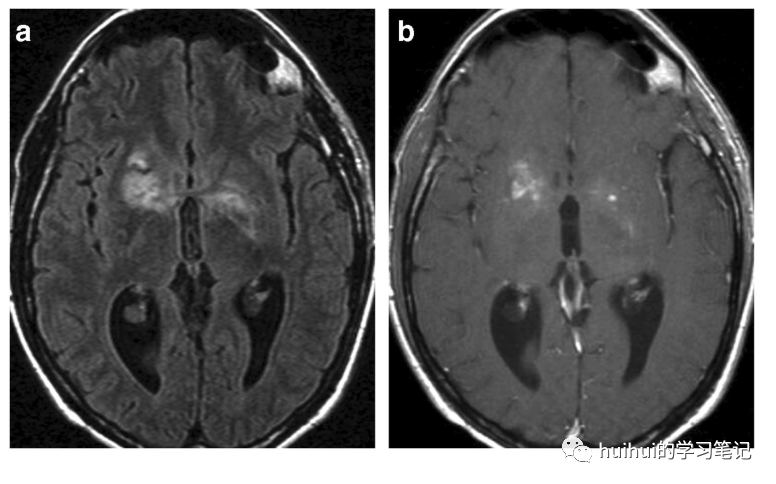
图18 隐球菌病。a 轴位FLAIR图像显示双侧基底神经节有不对称的斑块状高信号病变;b 在相应的对比后T1加权图像上这些病变内有局灶性强化区域
血管源性/缺血性
HIE经常涉及基底神经节和丘脑,因此在 "涉及基底神经节和丘脑的病理学 "一节中讨论
肿瘤性
生殖细胞肿瘤
颅内胚胎瘤主要发生在中线上的位置,主要是小脑上区和松果体区;然而,大约6-10%发生在非中线结构,通常是基底神经节(有时是丘脑,极少是后窝结构)。基底神经节胚胎瘤是出了名的难以诊断,因为它的临床表现没有特异性,在最初的影像学研究中也有细微的异常。病变可能是单侧或双侧的,在CT上仅显示轻微的高密度,同时在T1和T2加权MR图像上显示轻微的高信号,没有肿瘤占位效应或增强。如果这种细微的发现是双侧的,可能特别难以注意。SW-MRI(或gret2 *图像)特别有用,显示肿瘤苍白球和壳核明显低信号。另一方面,生殖细胞瘤可能伴有囊变、钙化或出血,甚至明显的肿块效应。同侧脑和/或脑干萎缩(图19和59),代表沃勒氏变性,发生于单侧病变的晚期。
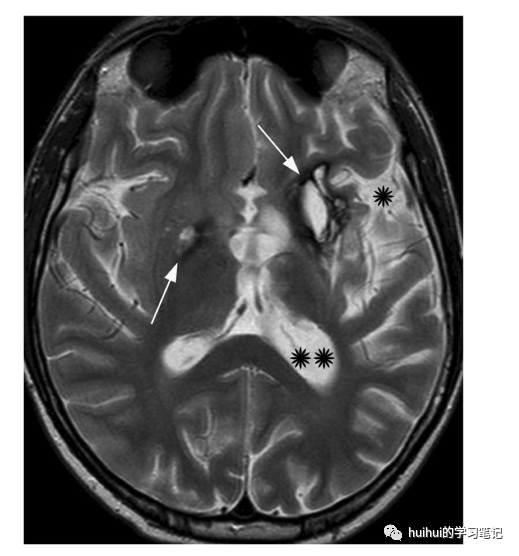
图19:生殖细胞瘤。一名14岁的男孩,认知和行为发生变化,头痛,右臂和右腿逐渐无力。轴向t2加权图像显示左侧基底节区高信号病灶大于右侧,周围低信号突出(箭头)。左侧偏瘫伴外侧裂(*)和侧脑室后角(**)增宽。
影响丘脑主要疾病
一、遗传性代谢/基因
溶酶体储存障碍
神经节苷脂沉积症(是一组由溶酶体酶缺乏引起的隐性遗传病。GM1和GM2(也称为泰-萨克斯病和山德霍夫病)被分为三种类型:婴儿慢性、青少年慢性和成人慢性。婴儿型是最常见和最严重的。患者在出生后的前6个月表现为快速发展的运动无力、发育迟缓、耳聋和失明,通常在1-3岁时死亡。青少年和成人慢性形式在运动障碍和智力障碍方面有不同的临床表现[41]。GM1亚型和GM2亚型儿童神经节神经节凹陷症患者的神经影像学表现相似。CT显示丘脑对称高密度,T1信号增高,T2信号降低(图20和图59)。其他特征包括轻度纹状体T2高信号和弥漫性髓磷脂减少,胼胝体保留。青少年和成人形态多表现为小脑萎缩。
其他溶酶体储存疾病也可表现为双侧丘脑CT高密度和MRI T2低强度,主要为Krabbe病和神经元蜡样脂褐素病。另一方面,“枕部征”表现为单侧或双侧丘脑枕部T1高强度,最初被认为是法布里病(FD)的常见和病理征象,但由于其发病率低(约3%的患者)和特异性低,它不再被认为是FD的特征性影像学表现。
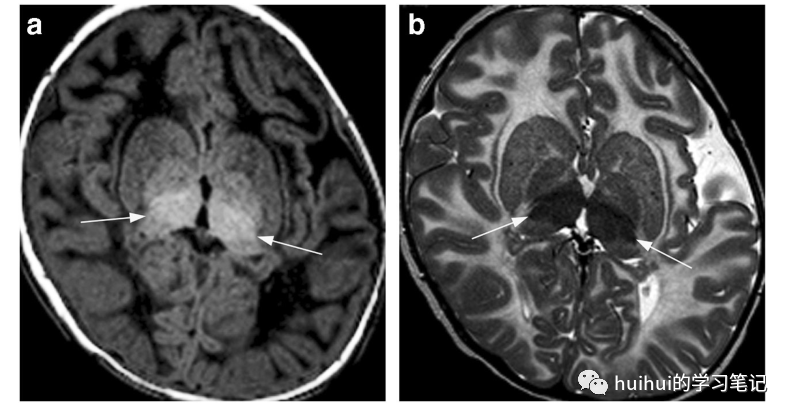
图20:溶酶体储存障碍- GM1和GM2神经节苷脂沉积症。一名17个月大的女婴,患有1型神经节苷脂沉积症的婴儿期。a t1加权像上双侧丘脑高信号(箭头)。b t2加权图像显示丘脑相应的低信号强度(箭头)。幕上白质弥漫性髓鞘减少(T1信号强度低,T2信号强度高)和斜头畸形
二、获得代谢/有毒
甲苯毒性/溶剂滥用
甲苯(甲苯)通常是工业溶剂和许多家庭溶剂的主要和最具毒性的产品,如胶水,汽油和指甲油。虽然接触可能是职业性的(涉及油漆的建筑工作、美甲沙龙的雇员和汽油生产),但娱乐上的滥用(“嗅胶”)已成为一个全球性的健康和社会问题。尽管甲苯诱发脑病的发病率很高,后果严重,但其毒性机制尚不清楚。
急性中毒的影像学检查通常不明显。MRI上的异常是在长期慢性滥用(至少一年,通常几年)后发现的,并且在检测时是不可逆的。特征性的发现是丘脑和其他灰质的低T2信号、皮质与皮质下白质的差的区分、模糊的白质T2高强度和整体脑萎缩(图21和59)。特征性的双侧丘脑低T2信号可能是由于伴有铁的继发性沉积的脱髓鞘和/或脂质膜内甲苯的直接积聚。滥用的持续时间与影像学发现的严重程度相关
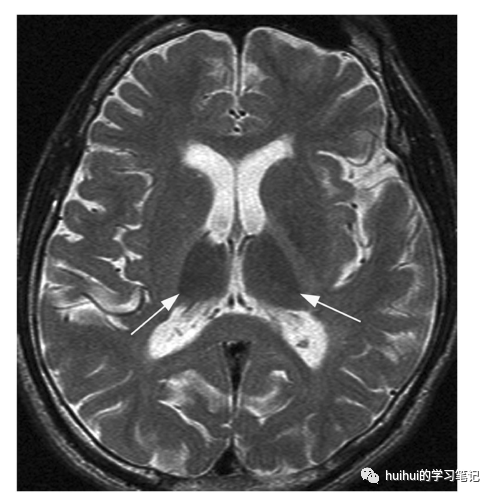
图21:甲苯毒性。21岁男性,有机溶剂滥用5年。轴向T2加权图像显示两侧丘脑明显低信号(箭头)。还可以看到左侧苍白球的信号强度轻度降低,全脑萎缩,白质信号弥漫性增加,失去了灰质-白质的区分。
Wernicke脑病
Wernicke脑病(WE)是一种由维生素B1(硫胺素)缺乏引起的急性、最初可逆的神经精神障碍,通常表现为眼部异常、共济失调和意识模糊。它通常发生在不良饮食环境中,典型的是酒精中毒,这是成人WE最常见的原因。然而,由胃肠道疾病和/或手术、妊娠剧吐和其他情况引起WE被认为是存在漏诊。尽管硫胺素摄入量不足是儿童长期营养不良的原因,但WE在患有恶性肿瘤和长期重症监护病房的儿科患者中越来越普遍。
MRI显示沿着内侧丘脑、中脑导水管周围灰质、四叠体板和乳头体的典型对称信号改变(图22、23和59)。在一些患者中,脊髓背侧、脑神经、小脑和中脑周围皮质也受到影响,典型的是非酒精性WE。病变在DWI呈高信号,FLAIR和T2加权像,在T1加权像上呈低信号或等信号,大约一半病例显示一定程度的增强。扩散率从降低到增加不等,并且可能是可逆的。WE是一种容易治疗的严重疾病,应该在MRI上进行疑诊和并进一步确诊。
甲硝唑诱导性脑病(MIE)是这种广泛使用的抗菌药物的一种罕见并发症,其临床表现可以模拟WE。甲硝哒唑毒性的可能机制之一是通过产生抑制性硫胺素类似物代谢物。特征性的磁共振表现是双侧小脑齿状核T2高信号,同时累及胼胝体压部、中脑导水管周围灰质和脑干背侧。中脑导水管周围灰质和中脑背侧的信号异常可能与WE难以区分;然而,乳头体和内侧丘脑的病变在MIE中极为罕见,而齿状核的病变在WE中相当罕见。有病例报道还描述了可能的WE和MIE组合的情况。
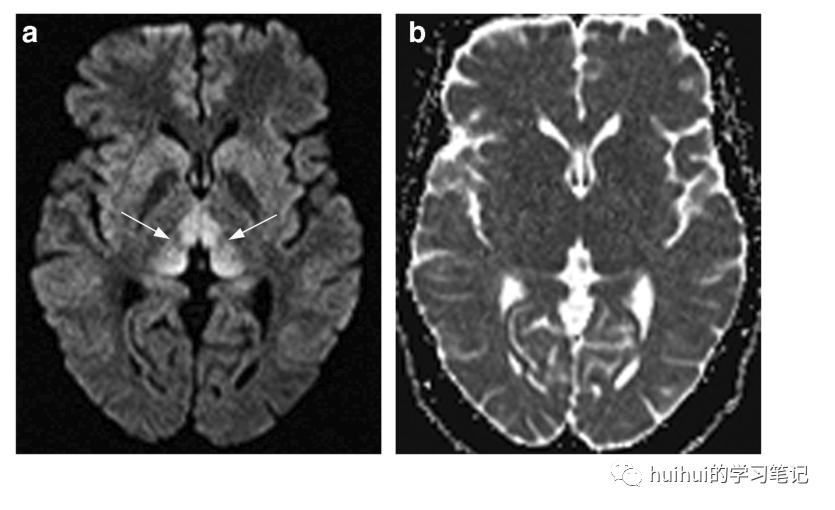
图23 Wernicke脑病,一名49岁男性因慢性酒精中毒而患WE。轴向DWI显示特征性的双侧对称丘脑背内侧高信号(箭头)。b .在相应的ADC图上没有扩散异常。
急性高氨血症脑病
急性高氨血症性脑病是一种威胁生命的疾病,最常见的原因是严重的肝病,但它也可能发生在非肝硬化的情况下,导致氨产生增加(如产尿素细菌的感染和多发性骨髓瘤)或氨清除减少-主要是尿素循环酶、门体分流和药物(如扑热息痛和丙戊酸钠)的遗传性疾病。患者表现为渐进性嗜睡和癫痫发作,继发于脑水肿和颅内高压。急性高氨血症性脑病可发展为昏迷和死亡,或导致长期后遗症,包括智力障碍。
典型的MRI发现是岛叶和扣带回皮质中的双侧对称信号异常,通常伴有丘脑受累(图24、25和59)。皮质和深灰色物质的受影响区域在T2加权图像上显示高信号,这在FLAIR和DWI上更引人注目,而在ADC图上可能出现扩散率降低。基底神经节通常会被保留下来,还有脑周皮质和枕叶皮质。
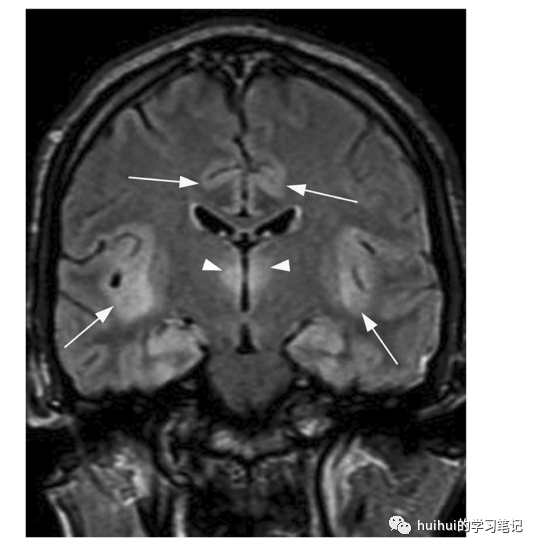
图24:急性高氨血症脑病。一名26岁男子被发现昏迷,检查时有上运动神经元体征、阵挛和嗜睡。生化检查显示由于丙戊酸钠毒性导致高氨血症。冠状面FLAIR图像显示双侧对称性高信号,包括岛叶和扣带回(箭头)、丘脑(箭头)和海马。
三、炎症和免疫
急性播散性脑脊髓炎(ADEM)患儿中,丘脑受累及白质病变较为常见。基底神经节也可能受到影响,因此ADEM将在“基底神经节和丘脑均受累的病理”一节中讨论。
急性坏死性脑炎(ANE)
一种与呼吸道病毒相关的脑炎亚型,表现为急性坏死性脑炎(ANE),一种典型累及双侧丘脑的快速进行性脑病。与ADEM相反,炎症反应不明显是典型特征,而坏死和出血是主要的发现。ANE之前通常有与病毒相关的发热性疾病,最常见的是流感,其他病原体包括冠状病毒疾病2019(新冠肺炎),随后迅速恶化。这种疾病主要(但不仅限于)发生在幼儿身上,也被称为儿童急性坏死性脑炎(ANEC)。ANE可能偶发;然而,存在与RANBP2基因突变相关的复发性和家族性形式。
非常典型的影像学发现是丘脑的双侧对称同心出血性病变,在SW-MRI/T2*图像上最明显,伴有周围水肿,通常伴有中央低和周围增加的扩散率(图26和59)。这种影像学表现也可以在深静脉血栓形成时看到;然而,ANE患者的大脑内静脉是通畅的。脑干和岛下区域,以及小脑和基底神经节也可能受到影响。
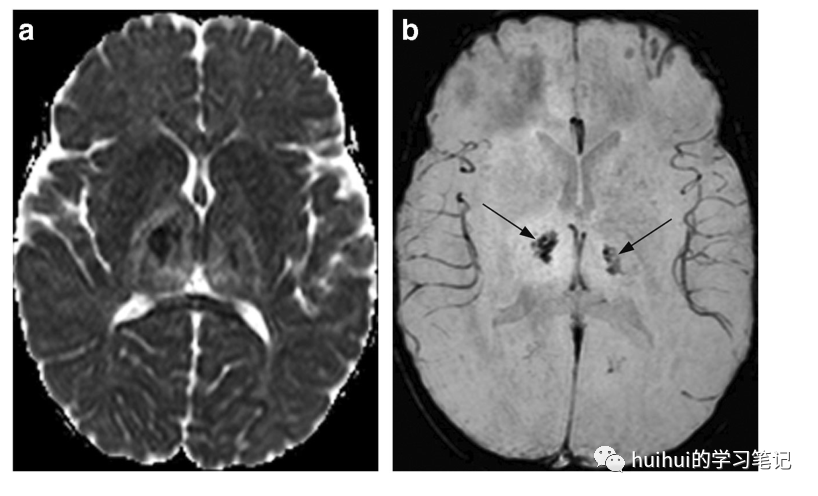
图26急性坏死性脑炎(ANE)。一名2岁女童因呼吸道病毒感染而嗜睡。轴向ADC图显示双侧丘脑肿胀,扩散性不均——主要是中央低信号和周围高信号。b:中央区域在相应的GRE T2*图像上显示信号丢失(箭头),与出血一致。大脑内静脉外观正常,无血栓形成迹象。
四、血管源性/缺血
Percheron动脉梗死
在大多数个体中,旁正中丘脑的动脉供应由来自两侧大脑后动脉(PCA)P1段的成对丘脑穿支动脉提供。然而,在少数情况下,这些穿通血管来自仅来自一个PCA的单一主干,称为Percheron动脉。这种变异与基底尖的不对称融合变异有关。这种血管的闭塞导致双侧丘脑梗死,累及或不累及吻侧中脑,心源性疾病被认为是最常见的原因。出现的急性症状包括视觉障碍、意识模糊和昏迷。
CT显示双侧丘脑旁正中低密度病变(图27a和59),通常是对称的。虽然CT最初可能是正常的,如其他急性梗死,但MRI更敏感,显示明亮的DWI信号,扩散率降低(图27 b和27c)和T2高信号。“中脑V征”是一个额外的影像学特征,在大多数患者中均有报道。它由沿脚间池中脑表面双侧延伸的FLAIR图像上的高信号强度组成。在慢性病变中发现具有CSF样密度、信号强度和扩散率的体积损失(图28)。
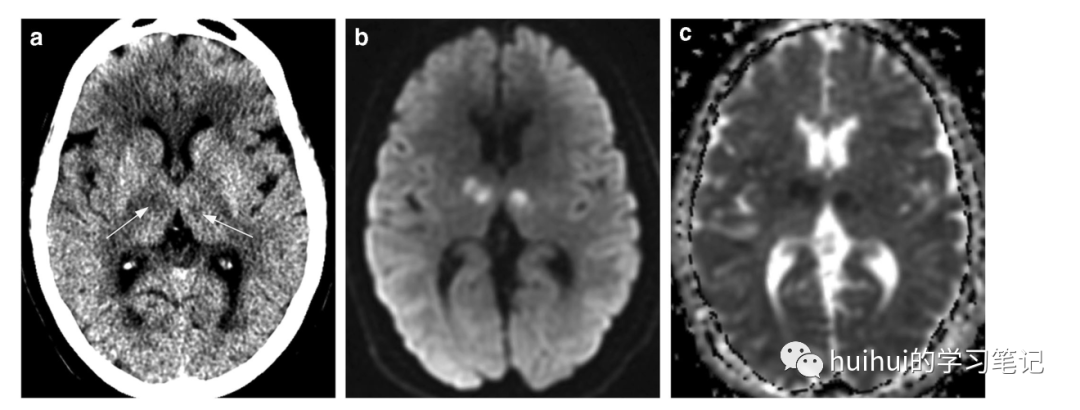
图27:Percheron动脉梗死。一名69岁的女性患者被发现昏迷。轴位非增强CT图像显示双侧丘脑的细微低密度病灶(箭头)。病变在DWI图上是亮的(b ),在ADC图上是暗的(c ),这与急性梗塞弥散率降低相一致
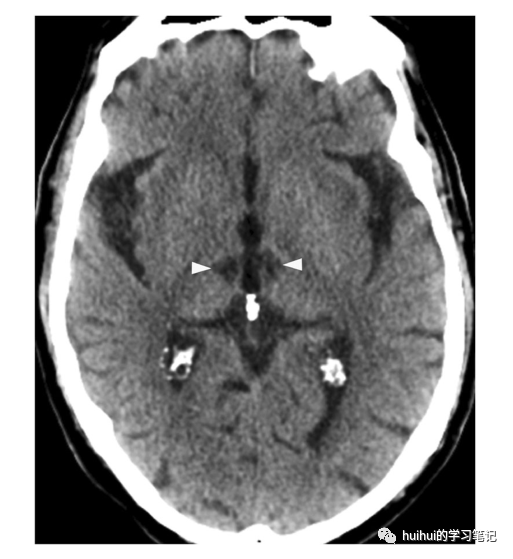
图28:Percheron动脉梗死。中央内侧丘脑边界清楚的双侧脑脊液样低密度病灶(箭头)代表慢性梗死。
颅内深静脉血栓形成
患有深脑静脉血栓形成(DCVT)的患者倾向于出现几天的头痛和意识减退史。DCVT可能是孤立的,也可能是更广泛的脑静脉血栓形成的一部分,由于缺乏实质性的侧支引流途径,它与比硬脑膜静脉血栓形成更糟糕的结果相关。两侧大脑内静脉血栓形成导致双侧丘脑病变,基底神经节、内囊、中脑和上小脑受到不同程度的受累。
在CT上,闭塞静脉的密度显著增高,丘脑(图29和59)和基底神经节(在某些情况下)的密度低且经常出血。根据血栓的时期和梗死的时期不同,MRI表现有所不同,通常是不均匀的,包括T2高信号和低信号,T1低信号和高信号(图30),以及DWI显示的弥散异常,弥散从高到低的变化。与动脉梗塞相反,观察到的信号异常主要是由于静脉充血和由此引起的血管源性水肿,而不是细胞毒性水肿。典型的病变是双侧的,然而在某些病例中明显不对称。
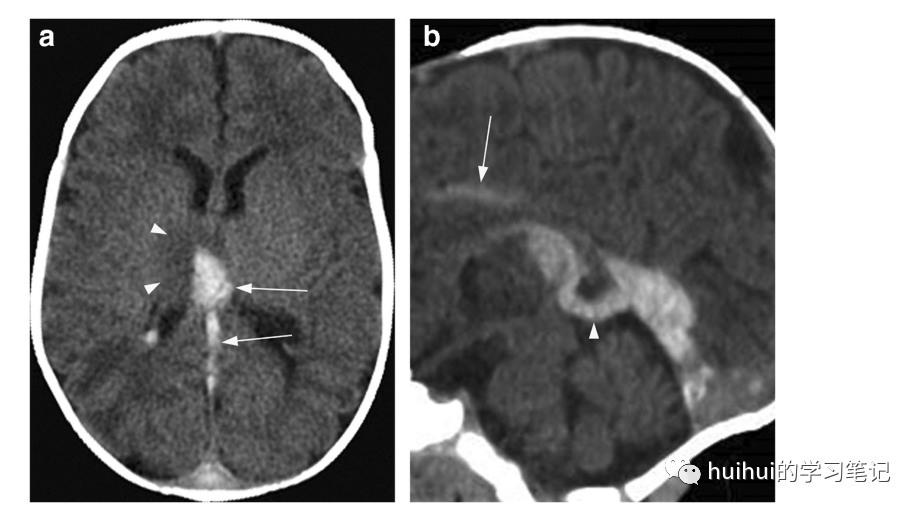
图29:深脑静脉血栓形成(DCVT)。一个意识丧失的6个月大的男孩。一个非增强的轴位CT图像显示右侧高于左侧的双侧丘脑高密度病灶,与血栓一致。右侧丘脑有轻度占位效应和周围低密度影(箭头所示)。血栓与延伸到Galen静脉的高密度区(箭头)相邻。重组的正中矢状面CT图像通过描绘大脑内静脉、Galen静脉(箭头)和直窦以及下矢状窦(箭头)的显著高密度。证实DCVT存在
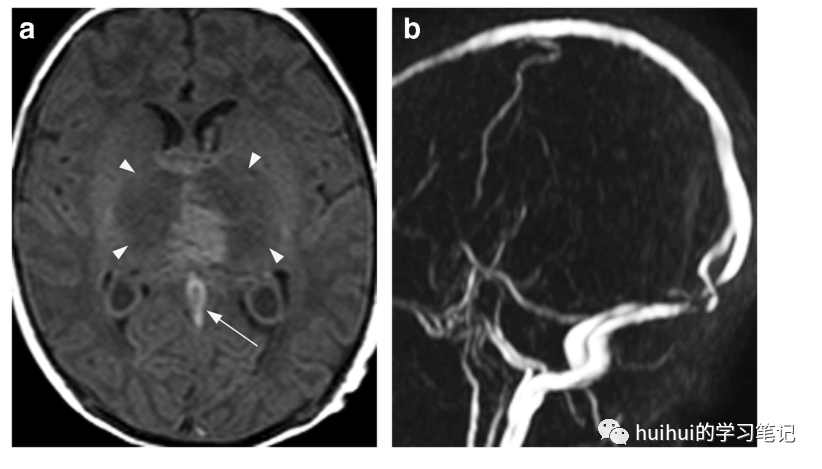
图30 DCVT。足月新生儿深静脉血栓形成。双侧丘脑肿胀,在轴位t1加权像上主要是低信号强度(箭头)。左侧高于右侧的中央高信号区是出血的征象。此外,在Galen静脉(箭头)有高信号强度,提示血栓形成。磁共振静脉成像显示大脑内静脉、盖伦静脉和直窦无信号,证实了DCVT
硬脑膜动静脉瘘/血管畸形
硬脑膜动静脉瘘(dAVF)的非出血性表现包括痴呆,这是由皮层静脉高血压或较少见的双侧丘脑静脉高血压引起的。dAVF引流入深静脉系统,主要是Galen静脉,导致双侧丘脑水肿和快速进行性痴呆
在CT上可以看到丘脑的轻度肿胀和减弱,而MRI显示双侧丘脑的T2信号强度和扩散性(ADC图上的高信号)弥漫性增加,并伴有轻度肿块效应。没有DCVT和出血迹象,尽管在SW-MRI上可能出现微出血。通过导管血管造影(脑血管DSA)确定诊断,然后可以进行血管内栓塞治疗。
五、肿瘤
弥漫性中线胶质瘤
在更新的世卫组织2016中枢神经系统肿瘤分类中,增加了一个新的实体,H3K27M突变体的弥漫性中线胶质瘤,指的是以前被指定为儿童弥漫性固有脑桥胶质瘤的肿瘤。进一步的研究表明,这种突变也发生在其他成人和儿童中线和旁正中肿瘤中,包括丘脑神经胶质瘤。丘脑的肿瘤性病变相对罕见,主要影响年轻人和儿童。定位较深和邻近关键结构意味着不完全切除或不可切除性和不良预后。最近发现双侧丘脑神经胶质瘤,与单侧相反,经常隐藏EGFR癌基因的突变,而H3K27M突变实际上是罕见的。此外,基因定制疗法(使用酪氨酸激酶抑制剂)在一些患有此类双侧眼睑肿瘤的儿科患者中显示出令人鼓舞的结果。
双侧和单侧丘脑胶质瘤在信号、扩散率和对比增强方面都有不同程度的改变,但在T2加权和FLAIR图像上的高信号是典型的。与本综述中描述的其他实体相比,突出的占位效应是它们的特征(图31和59)。
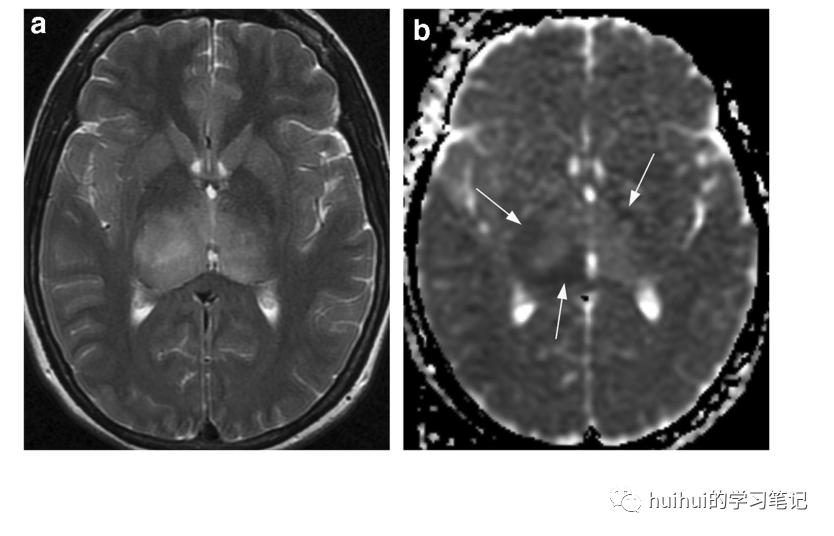
图31 双侧丘脑胶质瘤。44岁男性,头晕,左脸感觉异常,身体左侧无力。轴位T2加权像显示丘脑右侧大于左侧,信号强度轻度不均一性增加。相应的ADC图显示肿块病变的异质性,弥散率显著增加,周边减少(箭头所示)
原发性中枢神经系统淋巴瘤
原发性中枢神经系统淋巴瘤(PCNSL)最常见于深部脑结构和脑室周区域,通常接触脑脊液表面,并有沿室管膜扩散的趋势。内侧丘脑也可能受累,偶尔双侧对称。在某些情况下,病灶不接触脑脊液表面,基底神经节也可能受到影响。典型的影像学特征是均匀的轴内单发或多发边界清晰的肿块,在非增强CT上呈高密度(但与急性血凝块相比密度较低)(图32 ), T2信号低至等信号,周围血管源性水肿明显。病变特征性地表现为致密均匀强化和非常低的弥散,ADC图上呈现低信号(主要是由于细胞数目增多)。
虽然PCNSL的发病率在20世纪90年代中期略有下降,与艾滋病发病率的下降相一致,但它在免疫功能正常的老年人群中一直在增加。当在非增强CT扫描中沿脑室表面发现高密度的均匀肿块伴有明显的周围水肿时,应提高PCNSL的可能性。
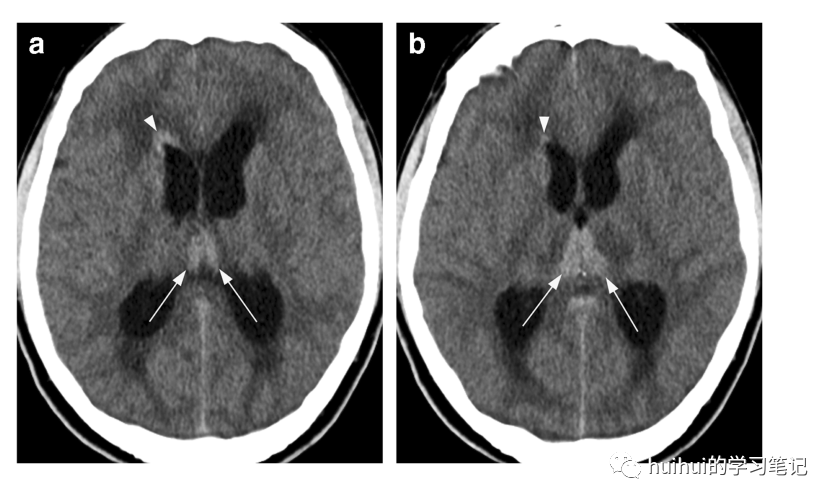
图32:原发性中枢神经系统淋巴瘤。一名46岁男性出现头痛、虚脱和呕吐。a、b轴位CT无增强图像显示丘脑内侧高密度灶(箭头),邻近低密度灶。另一个室管膜下高密度病变(箭头)位于右侧脑室前角。脑室扩张,脑室周围水肿,皮质沟消失,符合急性脑积水
同时累及基底神经节和丘脑的疾病
一、遗传代谢/遗传
微管蛋白病或微管蛋白相关的皮质发育不良
微管蛋白病是最近描述的一组具有广泛临床严重性的异质性脑畸形,由编码不同微管蛋白同种型的七种基因中的一种基因突变引起。微管蛋白和微管相关蛋白在皮质发育中起重要作用。微管蛋白病的诊断是基于MRI和基因检测中特征性脑畸形的存在。
深部灰质结构的形态异常是该疾病的标志(图33、34和59 ),是内囊白质组织异常产生的结果,如前内囊发育不全。在大多数情况下,基底神经节表现为大的圆形结构,其中尾状核、壳核和苍白球难以区分,丘脑肥大。在微侧裂脑亚型中,基底神经节要么不可见,要么严重发育不全。此外,微管蛋白病与脑干发育不全和小脑、大脑皮层和胼胝体的各种畸形有关。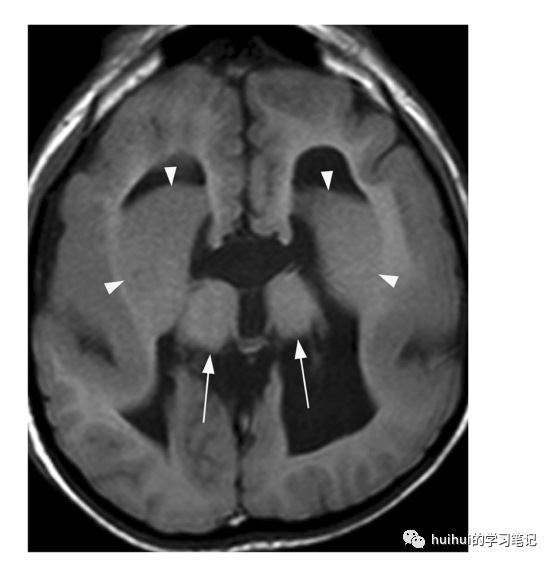
图33:微管蛋白病。一个10岁的智力障碍的女孩。在轴位t1加权像上,双侧基底节形态异常,尾状核和壳核(箭头)融合畸形。丘脑发育不全(箭头)。岛叶区域有巨脑回,同时缺少胼胝体、Z形脑干以及蚓部和小脑发育不全(未示出)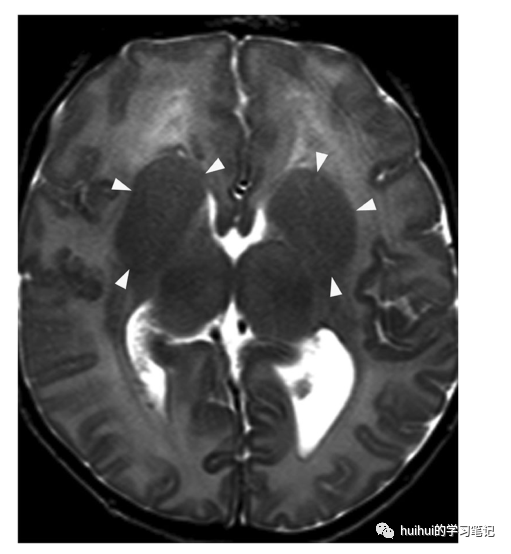
图34:微管蛋白病。一个1个月大的女孩发育不良。在轴位T2加权像上,基底神经节表现为尾状核和壳核融合的大圆形结构。丘脑稍微不对称
神经纤维瘤病1型
1型神经纤维瘤病(NF-1)是最常见的神经皮肤综合征/斑痣性错构瘤病。其典型表现包括咖啡斑、皮褶雀斑样斑、神经纤维瘤、视神经通路神经胶质瘤、错构瘤、Lisch结节(虹膜错构瘤)、特征性骨病变(如蝶骨发育不良)和动脉病变(如Moya-Moya综合征)
一个主要的神经影像学特征是局灶性异常信号(FASI),至少60–80%的儿童NF-1患者存在这种异常信号强度。这些是基底神经节、丘脑、脑干、小脑和偶尔大脑半球中分散的边界不清的T2高信号区,没有肿块效应(图35、36和59)。FASIs也可以在t1加权像上显示轻度高信号(图35b ),偶尔可以看到轻微的肿块效应。这些病变通常在CT上不可见,钆对比剂增强MRI很少出现增强。FASIs可能代表髓鞘空泡样改变的区域,但没有脱髓鞘的证据。这些发现的重要性尚不清楚,与认知功能障碍的因果关系仍有争议。目前还不清楚为什么这些变化会随着时间的推移而频繁退化,甚至完全消失,但在某些情况下也会重新出现或规模暂时增大。FASIs的主要鉴别诊断是低级别胶质瘤,通常表现为更实质性的肿块效应,在某些情况下,还表现为对比增强。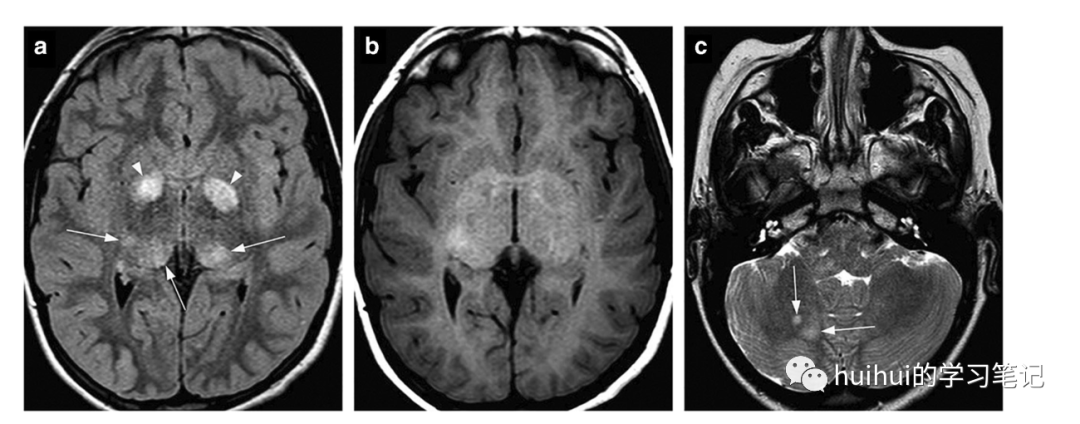
图35 NF-1的局灶性异常信号强度。一名8岁男孩经基因检测确诊为1型神经纤维瘤病。轴向FLAIR图像显示苍白球(箭头)和丘脑后部(箭头)斑片状双侧局灶性异常信号强度(FASIs)。在T1加权像上,一些病灶呈稍高信号。轴位T2加权像显示了小脑的额外FASIs(箭头)
原发性家族性脑钙化(Fahr病)
原发性家族性脑钙化(PFBC),以前被称为(家族性)特发性基底神经节钙化或Fahr病,是一种罕见的常染色体显性神经退行性疾病,以基底神经节和其他脑区双侧异常钙沉积为特征。这种疾病通常表现为异常运动、认知和精神表现的组合,在临床上与其他成人发病的神经退行性疾病难以区分。尽管PFBC在所有年龄组都有描述,但最常见于40或60岁发病。
除了基底神经节,丘脑(尤其是后外侧部分)、小脑齿状核和皮质下白质也经常受累,在CT上看得最清楚(图37和38)。在少数情况下,大脑和小脑皮质以及脑干也可能出现钙化。融合性脑白质异常也可能与之相关。继发性双侧钙化存在于各种遗传、发育、代谢、感染和其他疾病中。因此,对患有运动障碍、认知障碍和精神症状的患者进行评估的主要目的是排除代谢障碍,主要是甲状旁腺功能减退,这是继发性钙化的最常见原因,并找出该疾病的可能家族史。
具有类似颅内钙化模式的其他疾病包括Cockayne综合征和碳酸酐酶缺乏症-2型。Labrune综合征和Coats plus综合征显示致密岩石样钙化;但是,其还有白质信号改变,囊肿和对比增强等特征。
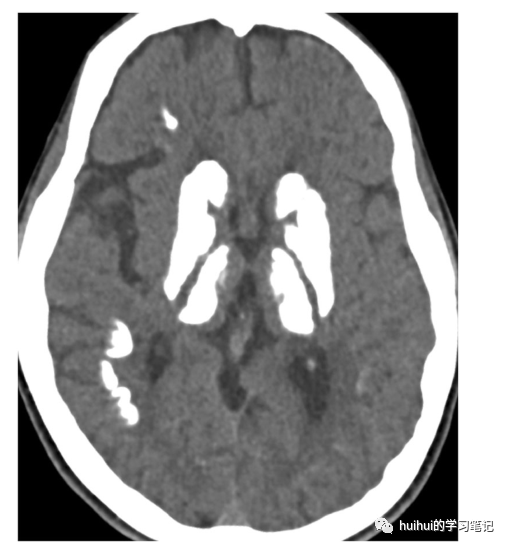
图37:原发性家族性脑钙化。一名30岁男性,患有原发性家族性脑钙化(PFBC)。轴位CT图像显示基底神经节、丘脑和皮质下白质广泛钙化。壳核出现萎缩。小脑齿状核也显示钙化(未显示)。

图38:原发性家族性脑钙化。一名57岁的PFBC男性。基底神经节(箭头)和丘脑(箭头)的钙化在矢状面t1加权像上主要是高信号
二、获得性代谢/毒性
乙二醇中毒
乙二醇(EG)是一种无色无味的液体,是防冻剂的主要成分。EG摄入通常是偶然的。其威胁生命的特征是由于乙二醇的代谢物与甲醇的代谢途径相同。乙醇酸的积聚导致代谢性酸中毒和随后的脑病。与甲醇相比,乙二醇摄入后12-24小时出现代谢效应。
大脑中央深部的对称性受累是急性EG中毒的特征性影像学表现(在最初几天内)。扩展到脑干和邻近大脑半球的双侧基底节和丘脑的弥漫性低密度,伴有轻度占位效应,是初次ct扫描的典型特征,然而,可能仍然不明显。MRI揭示了大脑中央基底部分内的双侧高T2信号(图39和59),主要在丘脑和基底神经节、海马和杏仁核,以及背侧中脑和脑桥。异常信号可延伸至小脑、脑干和脑岛的邻近部分,伴随着轻微的占位效应。还报告了双侧苍白球或壳核出血性病变,提示一氧化碳和甲醇中毒。
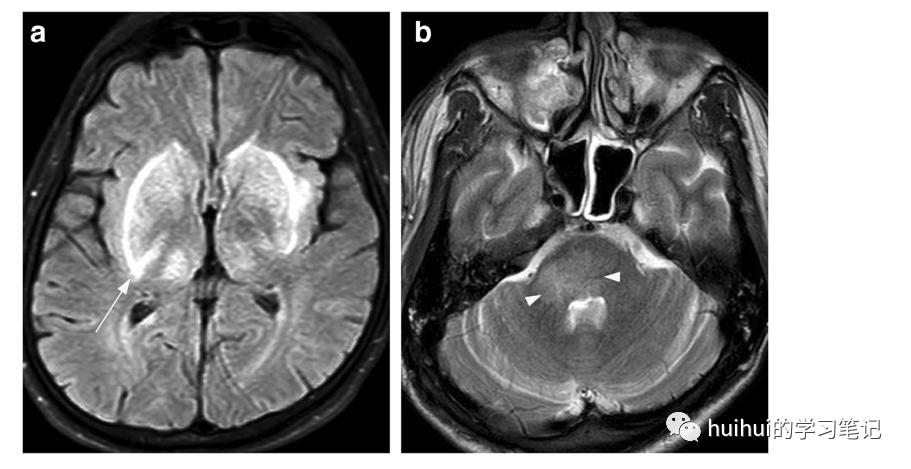
图39 乙二醇中毒。一名25岁的男性被发现在家中失去知觉,前一天晚上他的饮料中掺入了乙二醇(防冻剂)。最初的CT扫描并不明显(未显示)。两天后的MRI显示双侧对称肿胀,轴向FLAIR图像显示基底神经节和丘脑高信号。信号改变沿壳核的侧缘更明显,提示右侧有豆状叉征(箭头)。穿过脑桥的轴位T2加权像显示信号增强,延伸至右侧小脑中脚的脑干后部轻度肿胀(箭头所示)
渗透性脱髓鞘综合征
渗透性脱髓鞘综合征(ODS)通常与低钠血症的快速调整有关。然而,它可由各种电解质异常及其纠正引起,在慢性病导致虚弱的患者中经常遇到。ODS的组织学特征是髓鞘的非炎性破坏,同时保留了基本的轴突。该综合征的特征包括四肢瘫和在脑MRI上出现典型病变时的神经认知改变,代表脑桥中央髓鞘溶解症(CPM)和脑桥外髓鞘溶解症(EPM)。
然而,通常在症状出现后几天甚至几周会出现影像学特征的延迟,ADC图上扩散率的降低可能是最早的发现。EPM的标志是信号变化的显著对称性,这是代谢异常的一个特征,纹状体、丘脑、外囊和最外囊的T2高信号(图40a)。在CT上可以看到相应的低密度(图41)。海马体,小脑和大脑皮层也可能受累,弥散现象取决于扫描时间,EPM可以在没有或伴有CPM的情况下被观察到,影像学上表现为特征性的三叉戟形T2高信号区,在中央脑桥中具有低T1信号(图40b ),保留了皮质脊髓束。在某些情况下,EPM会先于CPM发生。
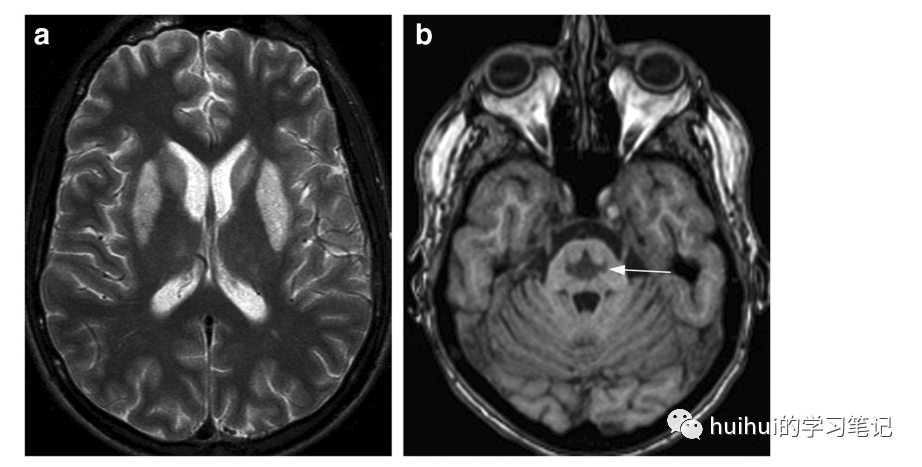
图40:渗透性脱髓鞘综合征(ODS)——脑桥外髓鞘溶解症(EPM)和脑桥中央髓鞘溶解症(CPM)。一名有酗酒背景的46岁男性出现低钠血症,该症状得到纠正,随后出现神经功能缺损加重,包括四肢瘫和意识丧失。基底神经节水平的轴向T2加权像显示壳核和尾状核双侧对称的高信号强度。两侧丘脑也有细微的高信号。穿过后颅窝的轴位T1加权像显示脑桥内呈“三叉戟”状的低信号病变(箭头),这是CPM的特征。
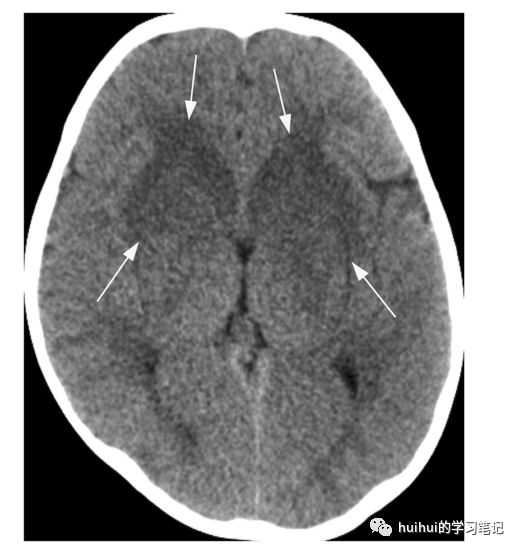 图41:脑桥外髓鞘溶解症(EPM)。一个有白血病背景的7岁女孩出现发热、呕吐和水平眼球震颤。入院时出现低钠血症,并及时调整。在非增强轴位CT图像上,以尾状头和前豆状核(箭头)为中心的基底神经节双侧对称肿胀和低密度。
图41:脑桥外髓鞘溶解症(EPM)。一个有白血病背景的7岁女孩出现发热、呕吐和水平眼球震颤。入院时出现低钠血症,并及时调整。在非增强轴位CT图像上,以尾状头和前豆状核(箭头)为中心的基底神经节双侧对称肿胀和低密度。
氨己烯酸治疗后的脑MRI改变(GABA-T抑制剂 ,增加脑内GABA含量。)
氨己烯酸,一种治疗婴儿痉挛的有效抗癫痫药物,在接受治疗的婴儿中,约有25%的婴儿出现脑部MRI异常。在丘脑中可以看到对称的T2高信号和弥散受限的特征性成像结果,这可能伴有苍白球、脑干被盖和小脑齿状核的受累(图42、43和59) 。这些可逆且通常完全无症状的发现与最大剂量的氨己烯酸有关。然而,在极少数情况下,可能会出现多动性运动障碍和危及生命的急性脑病。这种罕见的症状性MRI改变似乎不是剂量依赖性的,可能与伴随的激素治疗有关。在年龄较大的儿童和成人中尚未发现与氨己烯酸相关的MRI异常。
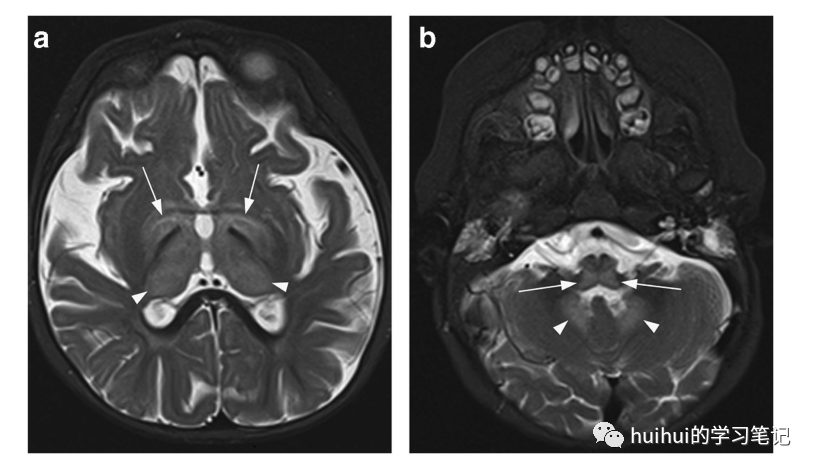
图42:氨己烯酸治疗的脑MRI变化。一名患有婴儿痉挛的50周大女孩接受了氨己烯酸治疗。轴位T2加权像显示丘脑(箭头)、基底神经节(箭头)、齿状核(箭头)和脑干(箭头)两侧对称高信号。没有相关的占位效应。这些发现是无症状的,并在停用氨己烯酸后完全消失。
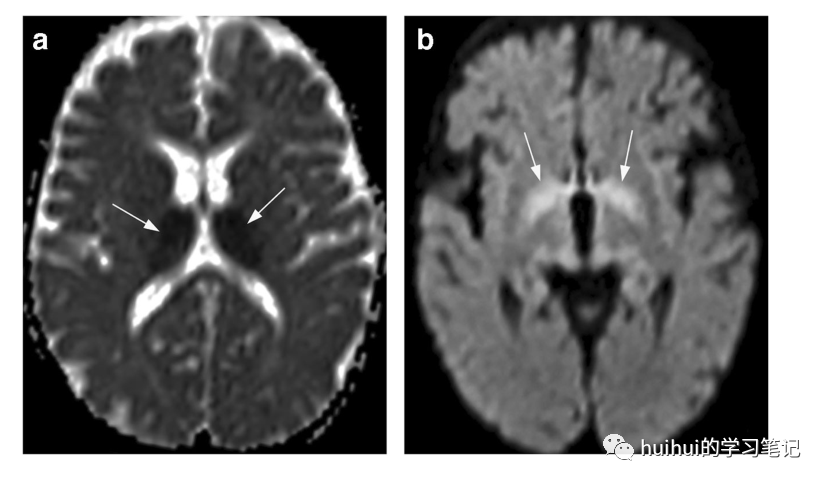
图43:氨己烯酸治疗的脑MRI变化。一名42周大的女孩因癫痫性脑病接受氨己烯酸治疗。a:轴向ADC图上低信号(箭头)的丘脑扩散率降低。低水平的DWI显示苍白球的高信号(箭头)。
与甲状旁腺激素紊乱相关的内分泌疾病
脑钙化分为三类:生理性、原发性和继发于钙代谢改变。显著的双侧颅内钙化最常见的病因是各种类型的甲状旁腺功能减退症和假性甲状旁腺功能减退症,它们可由许多疾病过程引起。在苍白球、壳核和尾状核的CT上观察到的双侧对称钙化,在T2加权和SWI磁共振上有相应的低信号,有时被称为Fahr综合征。丘脑、皮质下白质和小脑齿状核通常受影响的程度较小(图44),其次是大脑皮质、海马、皮质下和小脑白质。钙化的程度是可变的,取决于疾病的阶段和代谢异常的持续时间。原发性甲状旁腺功能减退症的相关发现是颅骨弥漫性斑片状溶骨性病变,而继发性甲状旁腺功能亢进症则出现斑块样硬脑膜钙化和血管钙化,最明显的是颈动脉钙化。
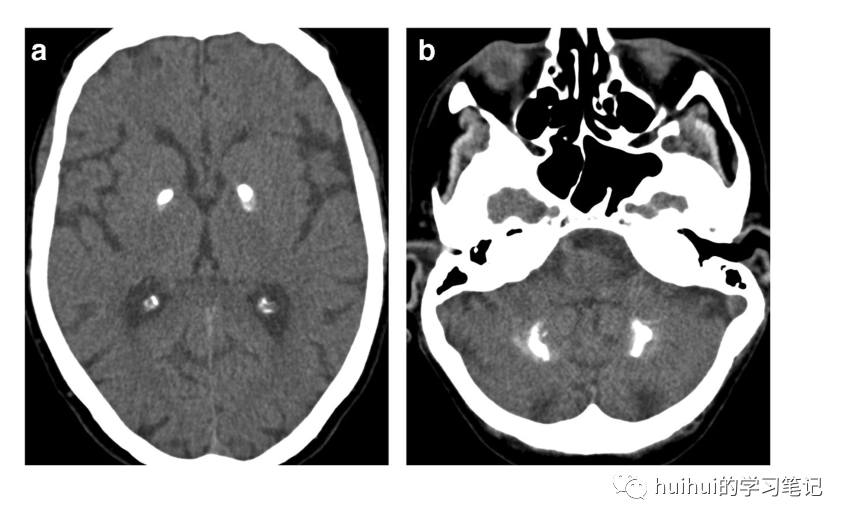
图44:甲状旁腺功能亢进。一名28岁男性出现新发震颤和甲状旁腺功能亢进,经实验室检查确诊。CT显示苍白球和齿状核内对称的密集钙化
三、克雅病(一大类疾病,与上述遗传、免疫等)
克雅氏病
克雅氏病(CJD)是一种快速进展的致命性神经退行性疾病,其典型特征是痴呆、共济失调、肌阵挛和行为改变。CJD是一种罕见的疾病,全球年发病率为1-2/100万,由神经元中异常形状的膜结合朊病毒蛋白积聚引起。其被细分为偶发性、家族性和获得性。大约85-90%的病例被归类为散发性CJD (sCJD ),其病因无法确定。遗传性或家族性CJD (fCJD)是由朊病毒蛋白基因的4种常见突变和许多其他突变引起的,占病例的10-15%。获得性(传染性)包括医源性克雅氏病和变异型克雅氏病(vCJD,源于食用受污染的牛肉引起的人畜共患病),占病例的不到1%。
DWI序列为其诊断是最有用的检查,准确率超过90%, 应该在所有快速进展的神经综合征患者中进行。这些发现包括FLAIR和DWI上的高信号,其局限于皮质(“皮质带状”)、纹状体(壳核和尾状核)、丘脑内侧和/或后侧或这些区域的组合中的灰质,而没有任何增厚或水肿(图45、46和59)。ADC图上经常出现低信号的扩散率下降,但这并不明显,可能会随时间而变化。除了双侧发现外,还经常观察到单侧皮质和深部灰质高信号(图59 ),这是一种典型的CJD模式(与代谢和毒性疾病中几乎总是对称的病变形成对比)。
sCJD的MRI标准是至少两个皮质区或尾状核和壳核的DWI信号异常。皮质DWI高信号似乎也是症状前的标志,因为MRI上的异常被发现早于sCJD的发病。FLAIR和DWI上双侧对称的后丘脑高信号被称为枕状征(伴随背内侧丘脑的受累,出现“曲棍球棒”样外观),被认为是vCJD的特征性表现,但只有当丘脑的信号比尾状核和壳核的信号更亮时才有意义。
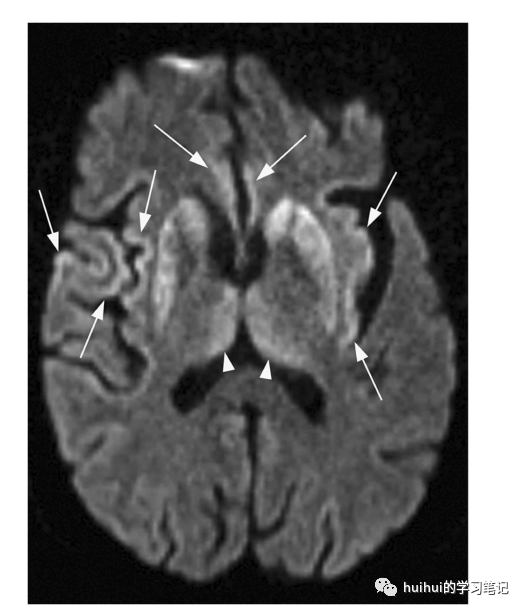
图45:克雅氏病(CJD)。一名69岁男性,有5个月的进行性全身无力、失语症和认知下降病史,并确诊为克雅病(CJD)。轴向DWI显示双侧纹状体和丘脑背内侧的高信号强度(“曲棍球棒”征,箭头)。在右侧脑盖、双侧岛叶和扣带回也有皮质高密度区(箭头所示)。
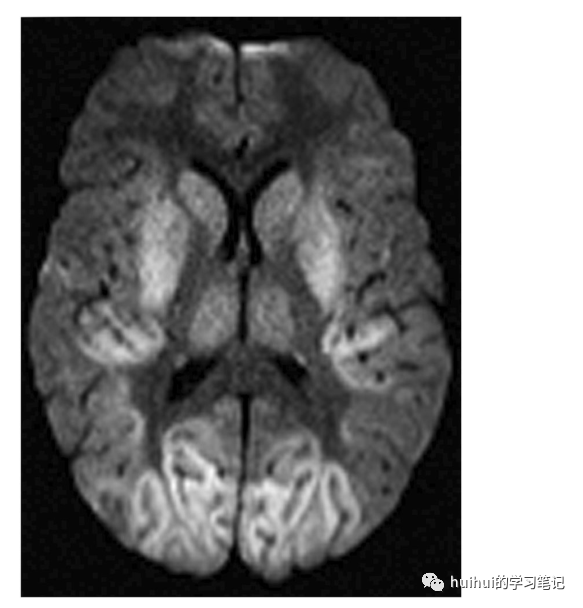
图46 CJD。一名61岁女性患有快速进行性痴呆、进行性运动和感觉障碍。确诊了CJD。轴DWI显示双侧基底节、丘脑和枕叶皮质高信号,部分颞叶皮质受累。
四、感染\炎症
急性播散性或脱髓鞘性脑脊髓炎
急性播散性脑脊髓炎(又名ADEM)是一种通常短暂、单相和自限性的免疫介导的炎性疾病,可累及大脑和脊髓,通常影响幼儿和青少年。基于交叉反应性,先前的病毒感染或较不频繁的免疫被认为触发了炎症过程。然而,许多病例没有明确的感染诱因史,而自身免疫性病因是可能的。在大约40%的病例中,发现了髓磷脂少突胶质细胞糖蛋白(MOG)的抗体。大多数患者完全康复,影像学检查结果也随之消失。在少数病例中,该疾病表现出多相病程或最终发展为多发性硬化。
在MRI上,除了白质(可能还有脊髓病变),基底神经节和丘脑经常双侧但不对称地受累,表现为边界不清的T2高信号病变(图47和59 ),这是一种可变的不均匀增强。病变的弥散率通常增加(图47b);然而,ADC图上周围暗带的低弥散区域并不少见,很可能对应于脱髓鞘的进展。这种病变可能很难与其他脱髓鞘疾病和传染性脑炎(选择性灰质受累)区分开来。与抗MOG抗体相关的ADEM也可能合并自身免疫性脑炎。
急性出血性脑脊髓炎(AHEM,Hurst病)是一种罕见而严重的疾病,以快速进行性肿胀白质病变和出血病灶为特征,在GRE T2*或SW-MRI上呈低信号,基底神经节和丘脑受累不一。伴有丘脑受累的AHEM偶尔会有与ANE非常相似的表现。
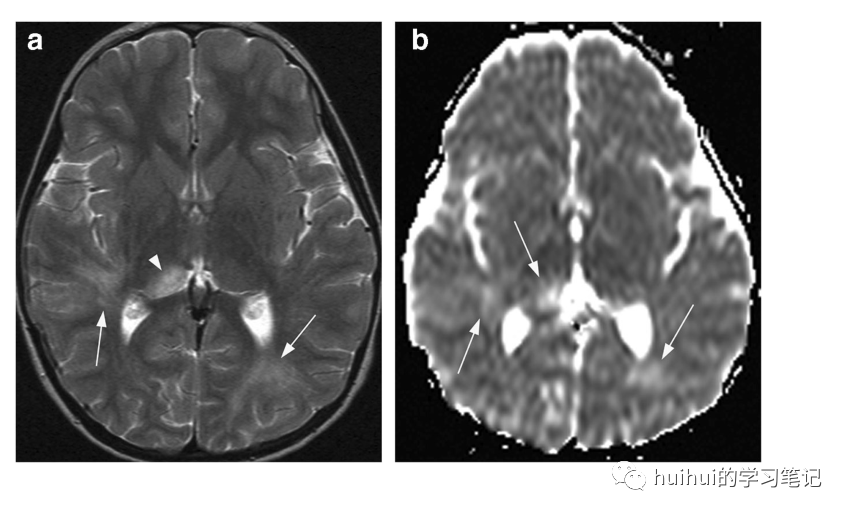
图47:ADEM。一名4岁女童出现行为改变和上呼吸道感染后癫痫发作。基底神经节水平的轴向T2加权像显示右侧丘脑背侧的局灶性高信号

